
33.4: Epidemiología Molecular
- Page ID
- 54815
La Epidemiología Molecular implica observar los biomarcadores moleculares de un estado de enfermedad. Esto incluye observar los perfiles de expresión génica, los patrones de metilación del ADN, es decir, la epigenómica y la estructura y organización de la cromatina en tipos celulares específicos. En capítulos anteriores, discutimos el vínculo entre la expresión génica (como ARN o proteínas) y los SNP en el contexto de estudios de eQTL. Como recordatorio, los EQTLs (loci de rasgos cuantitativos de expresión) buscan correlaciones lineales entre los niveles de expresión génica y diferentes variantes de un locus genético.
Esta sección se enfocará en comprender el papel de los marcadores epigenómicos como indicadores moleculares de una enfermedad. Es importante entender que múltiples factores, y por lo tanto múltiples conjuntos de datos entran en juego para comprender las bases epigenómicas de la enfermedad: patrones de metilación de pacientes de muestra (M), información genómica (G) para los mismos individuos, datos ambientales (E, cubriendo covariables como edad, género, tabaquismo hábitos etc.), y las cuantificaciones de fenotipos (P, pueden capturar múltiples marcadores fenotípicos, por ejemplo en la Enfermedad de Alzheimer, el número de placas neuronales por paciente). Además, necesitamos comprender las diversas interconexiones y dependencias entre estos conjuntos de datos para sacar conclusiones significativas sobre la influencia de la metilación para una determinada enfermedad.
Para eliminar covariantes experimentales, técnicas o ambientales, nos basamos en correcciones conocidas o inferidas por ICA (análisis de componentes pendientes). Para vincular los datos genéticos a los patrones de metilación, buscamos MeQTLs (loci trati cuantitativo de metilación), que es equivalente a los EQTLs. Los fenotipos moleculares como el nivel de expresión o el nivel de metilación también son rasgos cuantitativos. Finalmente, para vincular patrones de metilación con enfermedades, implementamos EWAS (Epigenome-wide association studies).
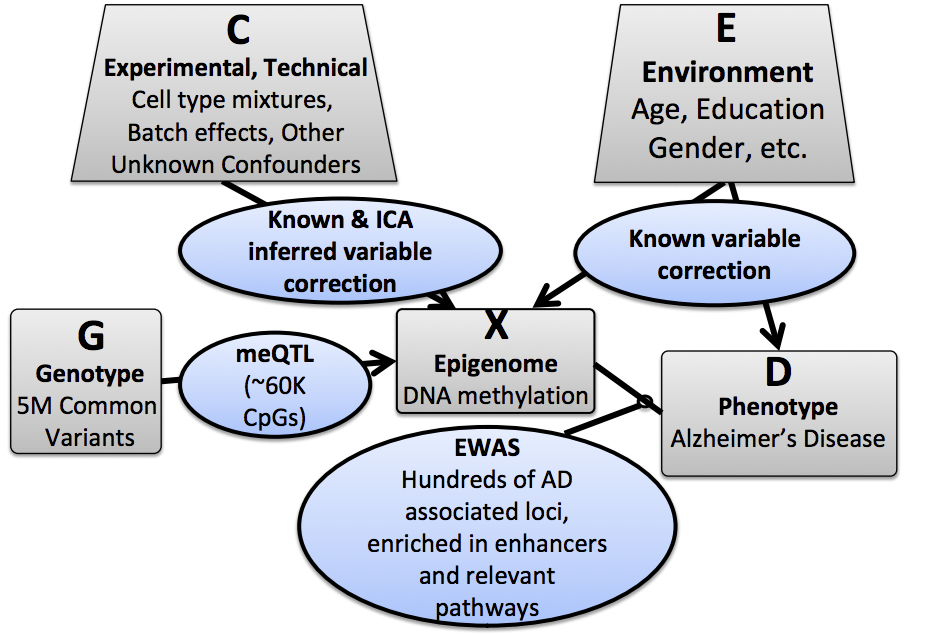
MEQTLS
El descubrimiento de MEQTLs sigue un proceso que es muy similar a la metodología utilizada para descubrir los EQTLs. Para descubrir cis-MEQTLs (es decir, MEQTLs donde el efecto sobre la metilación es proximal al locus probado) seleccionamos una ventana genómica, y utilizamos un modelo lineal para probar si vemos o no una correlación entre la metilación y las variantes de SNP en esa región. Probamos para ver si la correlación es significativa a través de una prueba F, donde nuestra hipótesis nula es que la complejidad del modelo adicional introducida a través de la información genómica no explica una porción significativa de variación en los patrones de metilación. Otros métodos para descubrir los MEQTLs incluyen la permutación y los modelos mixtos lineales (LMM).
Ejemplo Un ejemplo del uso de los MEQTLs para descubrir la conexión entre la metilación, el genotipo y la enfermedad es el Proyecto Memoria y Envejecimiento. 750 personas mayores inscritas en el proyecto hace muchos años y hoy en día, en su mayoría han muerto y han entregado su cerebro a la ciencia. Se determinó el genotipo y metilación de la corteza prefrontal lateral dorsal para estudiar la conexión entre la metilación y el fenotipo de Alzheimer y cómo el genotipo puede afectar el perfil de metilación. Se tomaron en cuenta los datos de SNP, metilación, factores ambientales (como edad, género, lote muestral, estado tabáquico, etc.) y fenotipo. Primeras covariantes necesitaron ser descubiertas y excluidas para asegurarse de que los resultados obtenidos no se deben a factores de confusión. Esto se hace descomponiendo la matriz de datos de metilación haciendo ICA. Esto permite el descubrimiento de variables que están impulsando la mayor variabilidad en el rasgo. La muestra discontinua y la mezcla celular pueden tener el mayor efecto en la variación entre individuos. Después de que esto se corrija, se utilizan modelos lineales, pruebas de permutación y modelos mixtos lineales para determinar cis-MEQTL, en qué medida el genotipo explica el nivel de metilación.
EWAS
Los estudios del genoma de todo el epigenoma (EWAS) tienen como objetivo encontrar conexiones entre el patrón de metilación de un paciente y su fenotipo. Al igual que GWAS, EWAS se basa en modelos lineales y pruebas de valor p para encontrar vínculos entre los perfiles epigenómicos y los estados de enfermedad. Junto con los MEQTLs, los EWAS también pueden arrojar luz sobre si un patrón de metilación dado es la causa o el resultado de una enfermedad. Idealmente, la idea es poder generar modelos que nos permitan predecir estados de enfermedad (fenotipos) basados en la metilación.
Hay algunos inconvenientes en EWAS. Primero, la varianza en los patrones de metilación por fenotipo suele ser muy pequeña, lo que dificulta la vinculación de estados epigenómicos con estados de enfermedad, similar a buscar una aguja en un pajar. Para mejorar esta situación, necesitamos controlar por otras fuentes de varianza en nuestros datos de metilación, como género, edad etc. El género, por ejemplo, incorpora una gran varianza para el caso de la Enfermedad de Alzheimer. Adicionalmente, necesitamos tener en cuenta la varianza por genotipo (en forma de MEQTLs). Además, la variabilidad entre las muestras es un problema importante en la recolección de datos de metilación para EWAS [? ]. Como diferentes tipos de células en un mismo individuo tendrán diferentes firmas epigenómicas, es importante que se colecten muestras de tejido relevantes y se corrijan los datos para los diferentes tipos de células/tejidos involucrados en un estudio.