
7: Alineación de Secuencias
- Page ID
- 117592

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)El programa de software BLAST (Basic Local Alignment Search Tool) utiliza algoritmos de alineación de secuencias para comparar una secuencia de consulta con una base de datos para identificar otras secuencias conocidas similares a la secuencia de consulta. A menudo, las anotaciones adjuntas a las secuencias ya conocidas arrojan información biológica importante sobre la secuencia de consulta. Casi todos los biólogos utilizan BLAST, haciendo de la alineación de secuencias uno de los algoritmos más importantes de la bioinformática.
La secuencia en estudio puede estar compuesta por nucleótidos (de los ácidos nucleicos ADN o ARN) o aminoácidos (de proteínas). Los ácidos nucleicos encadenan juntos cuatro nucleótidos diferentes: A, C, T, G para ADN y A, C, U, G para ARN; las proteínas encadenan veinte aminoácidos diferentes. La secuencia de una molécula de ADN o de una proteína es el orden lineal de nucleótidos o aminoácidos en una dirección especificada, definida por la química de la molécula. No es necesario que conozcamos los detalles exactos de la química; basta con saber que una proteína tiene extremos distinguibles llamados N-terminal y C-terminal, y que la convención habitual es leer la secuencia de aminoácidos desde el extremo N-terminal al C-terminal. La especificación de la dirección es más complicada para una molécula de ADN que para una molécula de proteína debido a la estructura de doble hélice del ADN, y esto se explicará en la Sección 7.1.
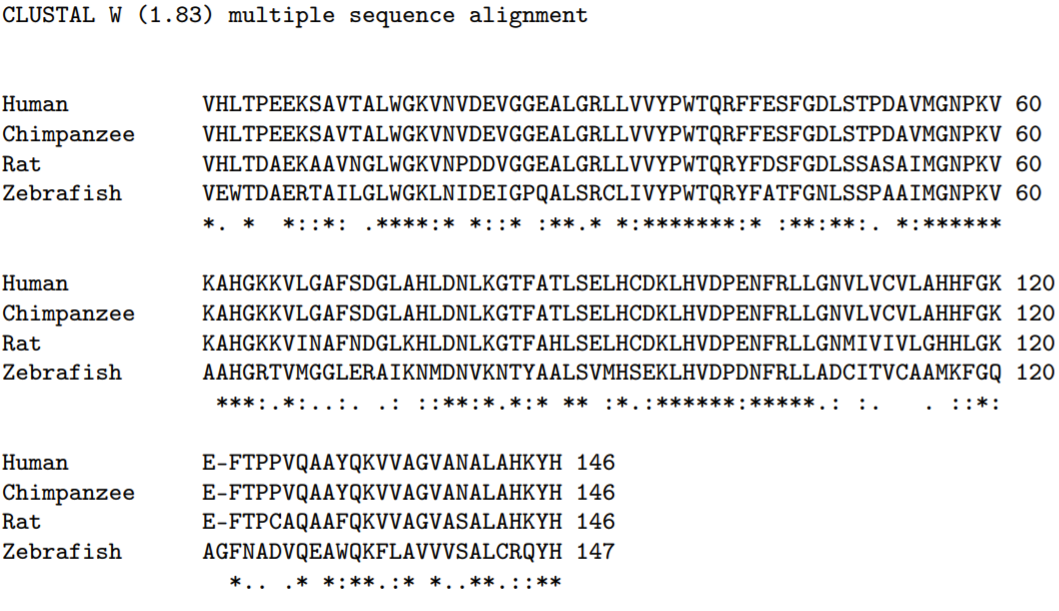
El algoritmo básico de alineación de secuencias alinea dos o más secuencias para resaltar su similitud, insertando un pequeño número de huecos en cada secuencia (generalmente denotados por guiones) para alinear siempre que sea posible caracteres idénticos o similares. Por ejemplo, la Fig\(7.1\) presenta una alineación usando la herramienta de software ClustalW de la cadena beta de hemoglobina de un ser humano, un chimpancé, una rata y un pez cebra. Las secuencias humana y chimpancé son idénticas, consecuencia de nuestra estrecha relación evolutiva. La secuencia de rata difiere de humano/chimpancé en solo 27 de 146 aminoácidos; todos somos mamíferos. La secuencia del pez cebra, aunque claramente relacionada, diverge significativamente. Observe la inserción de un hueco en cada una de las secuencias de mamíferos en la posición 122 del aminoácido pez cebra. Esto permite que la secuencia posterior del pez cebra se alinee mejor con las secuencias de los mamíferos, e implica ya sea una inserción de un nuevo aminoácido en peces, o una deleción de un aminoácido en mamíferos. La inserción o deleción de un carácter en una secuencia se denomina indel. Los desapareamientos en la secuencia, como el que ocurre entre el pez cebra y los mamíferos en las posiciones de los aminoácidos 2 y 3, se denomina mutación. ClustalW coloca un “* en la última línea para denotar coincidencias exactas de aminoácidos en todas las secuencias,\(\mathrm{a}^{\prime}: '\) y '\('\)' para denotar aminoácidos químicamente similares en todas las secuencias (cada aminoácido tiene propiedades químicas características, y los aminoácidos se pueden agrupar de acuerdo a propiedades similares). En este capítulo, detallamos los algoritmos utilizados para alinear secuencias.
- 7.2: Alineación de Fuerza Bruta
- Un enfoque (malo) para la alineación de secuencias es alinear las dos secuencias de todas las formas posibles, puntuar las alineaciones con un sistema de puntuación supuesto y determinar la alineación de mayor puntuación. El problema con este enfoque de fuerza bruta es que el número de alineamientos posibles crece exponencialmente con la longitud de la secuencia; y para secuencias de longitud razonable, el cálculo ya es imposible.
- 7.3: Programación dinámica
- Dos secuencias de tamaño razonable no pueden alinearse por la fuerza bruta. Por suerte, hay otro algoritmo prestado de la informática, la programación dinámica, que hace uso de una matriz dinámica.
- 7.4: Brechas
- La evidencia empírica sugiere que las brechas se agrupan, tanto en secuencias de nucleótidos como de proteínas. La agrupación suele ser modelada por diferentes penalizaciones por apertura de brechas.
- 7.5: Alineaciones locales
- Hasta ahora hemos discutido cómo alinear dos secuencias en toda su longitud, llamadas alineamiento global. A menudo, sin embargo, es más útil alinear dos secuencias en solo una parte de sus longitudes, lo que se denomina alineación local. En bioinformática, el algoritmo para la alineación global se llama “Needleman-Wunsch”, y el algoritmo para la alineación local “Smith-Waterman”.
- 7.6: Software
- Si tienes a mano dos o más secuencias que te gustaría alinear, hay una selección de herramientas de software disponibles.