28.4: Cromatografía de Reparto
- Page ID
- 79031

De las muchas formas de cromatografía líquida, la cromatografía de partición es la más común. En la cromatografía de reparto, el tiempo de retención de un soluto está determinado por la medida en que se mueve de la fase móvil a la fase estacionaria, y de la fase estacionaria de nuevo a la fase móvil. La extensión de esta partición en equilibrio está determinada por la polaridad de los solutos, la fase estacionaria y la fase móvil. En la cromatografía de partición de fase normal, la fase estacionaria es polar y la fase móvil es no polar (o de baja polaridad), con solutos más polares que tardan más en eluirse ya que son retenidos más fuertemente por la fase estacionaria polar. En la cromatografía de partición de fase inversa, la fase estacionaria es no polar y la fase móvil es polar, con solutos más polares eluyendo más rápidamente ya que son retenidos menos fuertemente por la fase estacionaria. De los dos modos, la cromatografía de partición en fase inversa es la más común.
Fases estacionarias para cromatografía de partición
En la cromatografía de partición, la fase estacionaria es una película líquida recubierta sobre un material de empaque, típicamente partículas de sílice porosas de 3—10 μm. Debido a que la fase estacionaria puede ser parcialmente soluble en la fase móvil, puede eluirse o sangrar de la columna con el tiempo. Para evitar la pérdida de fase estacionaria, lo que acorta la vida útil de la columna, se une covalentemente a las partículas de sílice. Las fases estacionarias unidas se crean haciendo reaccionar las partículas de sílice con un organoclorosilano de la forma general Si (CH 3) 2 RCl, donde R es un grupo alquilo o alquilo sustituido.
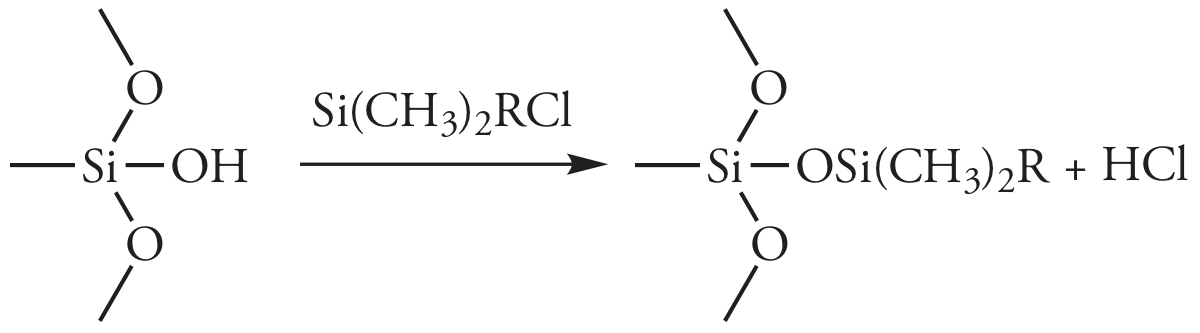
Para evitar interacciones no deseadas entre los solutos y cualquier grupo —SiOH restante, se usa Si (CH 3) 3 Cl para convertir sitios sin reaccionar en\(–\text{SiOSi(CH}_3)_3\); tales columnas se designan como terminalmente rematadas.
Las propiedades de una fase estacionaria dependen del grupo alquilo del organosilano. Si R es un grupo funcional polar, entonces la fase estacionaria es polar. Ejemplos de fases estacionarias polares incluyen aquellas en las que R contiene un grupo funcional ciano (—C 2 H 4 CN), un diol (—C 3 H 6 OCH 2 CHOHCH 2 OH) o un grupo funcional amino (—C 3 H 6 NH 2). Las fases estacionarias no polares más comunes utilizan un organoclorosilano donde el grupo R es una cadena hidrocarbonada n-octilo (C 8) o n-octildecilo (C 18). La mayoría de las separaciones de fase inversa se llevan a cabo usando una solución acuosa tamponada como fase móvil polar, o usando otros disolventes polares, como metanol y acetonitrilo. Debido a que el sustrato de sílice puede sufrir hidrólisis en soluciones básicas, el pH de la fase móvil debe ser inferior a 7.5.
Parece extraño que la forma más común de cromatografía líquida se identifique como fase inversa en lugar de fase normal. Uno de los primeros ejemplos de cromatografía fue la separación de pigmentos vegetales por parte de Mikhail Tswett, que utilizó una columna polar de carbonato de calcio y una fase móvil no polar de éter de petróleo. La asignación de normal e invertida, por lo tanto, tiene que ver con la precedencia.
Fases móviles para cromatografía de partición
El orden de elución de los solutos en HPLC se rige por la polaridad. Para una separación de fase normal, un soluto de menor polaridad pasa proporcionalmente menos tiempo en la fase estacionaria polar y eluye ante un soluto que es más polar. Dada una fase estacionaria particular, los tiempos de retención en HPLC de fase normal se controlan ajustando las propiedades de la fase móvil. Por ejemplo, si la resolución entre dos solutos es pobre, el cambio a una fase móvil menos polar mantiene los solutos en la columna por más tiempo y brinda más oportunidades para su separación. En HPLC de fase inversa el orden de elución es el opuesto al de una separación de fase normal, con solutos más polares eluyendo primero. El aumento de la polaridad de la fase móvil conduce a tiempos de retención más largos. Los tiempos de retención más cortos requieren una fase móvil de menor polaridad.
Elección de una Fase Móvil: Uso del Índice de Polaridad
Existen varios índices que ayudan a seleccionar una fase móvil, uno de los cuales es el índice de polaridad [Snyder, L. R.; Glajch, J. L.; Kirkland, J. J. Practical HPLC Method Development, Wiley-Inter- science: New York, 1988]. El cuadro 28.4.1 proporciona valores del índice de polaridad\(P^{\prime}\), para varias fases móviles comunes, donde valores mayores de\(P^{\prime}\) corresponden a disolventes más polares. Mezclar dos o más fases móviles, asumiendo que son miscibles, crea una fase móvil de polaridad intermedia. Por ejemplo, una fase móvil binaria preparada combinando el disolvente A y el disolvente B tiene un índice de polaridad\(P_{AB}^{\prime}\), de
\[P_{A B}^{\prime}=\Phi_{A} P_{A}^{\prime}+\Phi_{B} P_{B}^{\prime} \label{12.1} \]
donde\(P_A^{\prime}\) y\(P_B^{\prime}\) son los índices de polaridad para los solventes A y B, y\(\Phi_A\) y\(\Phi_B\) son las fracciones de volumen para los dos solventes.
| fase móvil | índice de polaridad (\(P^{\prime}\)) | Corte UV (nm) |
|---|---|---|
| ciclohexano | \ (P^ {\ prime}\)) ">0.04 | 210 |
| n-hexano | \ (P^ {\ prime}\)) ">0.1 | 210 |
| tetracloruro de carbono | \ (P^ {\ prime}\)) ">1.6 | 265 |
| i-propil éter | \ (P^ {\ prime}\)) ">2.4 | 220 |
| tolueno | \ (P^ {\ prime}\)) ">2.4 | 286 |
| éter dietílico | \ (P^ {\ prime}\)) ">2.8 | 218 |
| tetrahidrofurano | \ (P^ {\ prime}\)) ">4.0 | 220 |
| etanol | \ (P^ {\ prime}\)) ">4.3 | 210 |
| acetato de etilo | \ (P^ {\ prime}\)) ">4.4 | 255 |
| dioxano | \ (P^ {\ prime}\)) ">4.8 | 215 |
| metanol | \ (P^ {\ prime}\)) ">5.1 | 210 |
| acetonitrilo | \ (P^ {\ prime}\)) ">5.8 | 190 |
| agua | \ (P^ {\ prime}\)) ">10.2 | — |
Se realiza una separación por HPLC de fase inversa utilizando una fase móvil de 60% v/v de agua y 40% v/v de metanol. ¿Cuál es el índice de polaridad de la fase móvil?
Solución
Usando la Ecuación\ ref {12.1} y los valores de la Tabla 28.4.1 , el índice de polaridad para una mezcla de 60:40 agua-metanol es
\[P_{A B}^{\prime}=\Phi_\text{water} P_\text{water}^{\prime}+\Phi_\text{methanol} P_\text{methanol}^{\prime} \nonumber \]
\[P_{A B}^{\prime}=0.60 \times 10.2+0.40 \times 5.1=8.2 \nonumber \]
Supongamos que necesita una fase móvil con un índice de polaridad de 7.5. Explica cómo puedes preparar esta fase móvil usando metanol y agua.
- Contestar
-
Si dejamos que x sea la fracción de agua en la fase móvil, entonces 1 — x es la fracción de metanol. Sustituyendo estos valores en la Ecuación\ ref {12.1} y resolviendo para x
\[7.5=10.2 x+5.1(1-x) \nonumber \]
\[7.5=10.2 x+5.1-5.1 x \nonumber \]
\[2.4=5.1 x \nonumber \]
da x como 0.47. La fase móvil es 47% v/v de agua y 53% v/v de metanol.
Como regla general, un cambio de dos unidades en el índice de polaridad corresponde a un cambio de aproximadamente 10 veces en el factor de retención de un soluto. Aquí hay un ejemplo sencillo. Si el factor de retención de un soluto, k, es 22 cuando se usa agua como fase móvil (\(P^{\prime}\)= 10.2), entonces cambiar a una fase móvil de 60:40 agua-metanol (\(P^{\prime}\)= 8.2) disminuye k a aproximadamente 2.2. Tenga en cuenta que el factor de retención se vuelve más pequeño porque estamos cambiando de una fase móvil más polar a una fase móvil menos polar en una separación de fase inversa.
Elección de una fase móvil: Ajuste de la selectividad
Cambiar el índice de polaridad de la fase móvil cambia el factor de retención de un soluto. Como aprendimos en el Capítulo 26.4, sin embargo, un cambio en k no es una manera efectiva de mejorar la resolución cuando el valor inicial de k es mayor que 10. Para efectuar una mejor separación entre dos solutos debemos mejorar el factor de selectividad,\(\alpha\). Existen dos métodos comunes para aumentar\(\alpha\): agregar un reactivo a la fase móvil que reacciona con los solutos en una reacción de equilibrio secundario o cambiar a una fase móvil diferente.
Aprovechar una reacción de equilibrio secundario es una estrategia útil para mejorar una separación [(a) Foley, J. P. Chromatography, 1987, 7, 118—128; (b) Foley, J. P.; May, W. E. Anal. Chem. 1987, 59, 102—109; (c) Foley, J. P.; May, W. E. Anal. Chem. 1987, 59, 110—115]. La Figura 28.4.1 muestra la separación en fase inversa de cuatro ácidos débiles —ácido benzoico, ácido tereftálico, ácido p-aminbenzoico y ácido p-hidroxibenzoico — en una columna C 18 no polar usando un tampón acuoso de ácido acético y acetato de sodio como fase móvil. Los tiempos de retención para estos ácidos débiles son más cortos cuando se usa una fase móvil menos ácida porque cada soluto está presente en una forma de base débil aniónica que es menos soluble en la fase estacionaria no polar. Si el pH de la fase móvil es suficientemente ácido, los solutos están presentes como ácidos débiles neutros que son más solubles en la fase estacionaria y tardan más en eluirse. Debido a que los solutos ácidos débiles no tienen valores idénticos de p K a, el pH de la fase móvil tiene un efecto diferente en el tiempo de retención de cada soluto, lo que nos permite encontrar el pH óptimo para efectuar una separación completa de los cuatro solutos.
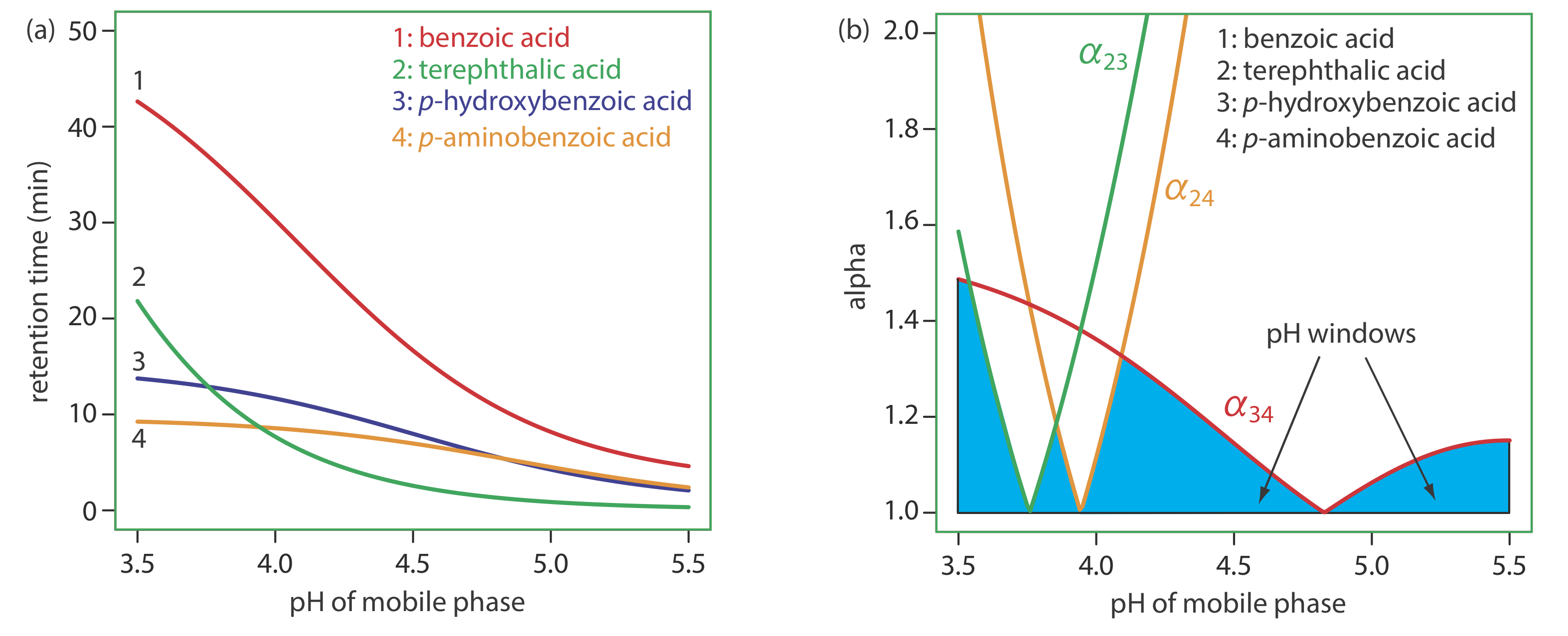
En Ejemplo 28.4.1 aprendimos a ajustar la polaridad de la fase móvil mezclando dos disolventes. Un índice de polaridad, sin embargo, es solo una guía, y las mezclas binarias de fase móvil con índices de polaridad idénticos pueden no resolver por igual un par de solutos. La Tabla 28.4.2 , por ejemplo, muestra los tiempos de retención para cuatro ácidos débiles en dos fases móviles con valores casi idénticos para\(P^{\prime}\). Aunque el orden de elución es el mismo para ambas fases móviles, el tiempo de retención de cada soluto se ve afectado de manera diferente por la elección del disolvente orgánico. Si cambiamos de usar acetonitrilo a tetrahidrofurano, por ejemplo, encontramos que el ácido benzoico eluye más rápidamente y que el ácido p-hidroxibenzoico eluye más lentamente. Aunque podemos resolver completamente estos dos solutos usando una fase móvil que es 16% v/v acetonitrilo, no podemos resolverlos si la fase móvil es 10% tetrahidrofurano.
| tiempo de retención (min) |
16% acetonitrilo (CH 3 CN) 84% pH 4.11 tampón acuoso (\(P^{\prime}\)= 9.5) |
10% tetrahidrofurano (THF) Tampón acuoso al 90% pH 4.11 (\(P^{\prime}\)= 9.6) |
|---|---|---|
| \(t_\text{r, BA}\) | \ (P^ {\ prime}\) = 9.5) ">5.18 | \ (P^ {\ prime}\) = 9.6) ">4.01 |
| \(t_\text{r, PH}\) | \ (P^ {\ prime}\) = 9.5) ">1.67 | \ (P^ {\ prime}\) = 9.6) ">2.91 |
| \(t_\text{r, PA}\) | \ (P^ {\ prime}\) = 9.5) ">1.21 | \ (P^ {\ prime}\) = 9.6) ">1.05 |
| \(t_\text{r, TP}\) | \ (P^ {\ prime}\) = 9.5) ">0.23 | \ (P^ {\ prime}\) = 9.6) ">0.54 |
| \ (P^ {\ prime}\) = 9.6)” class="lt-chem-362587">
Clave: BA es ácido benzoico; PH es ácido p-hidroxibenzoico; PA es ácido p-aminbenzoico; TP es ácido tereftálico |
||
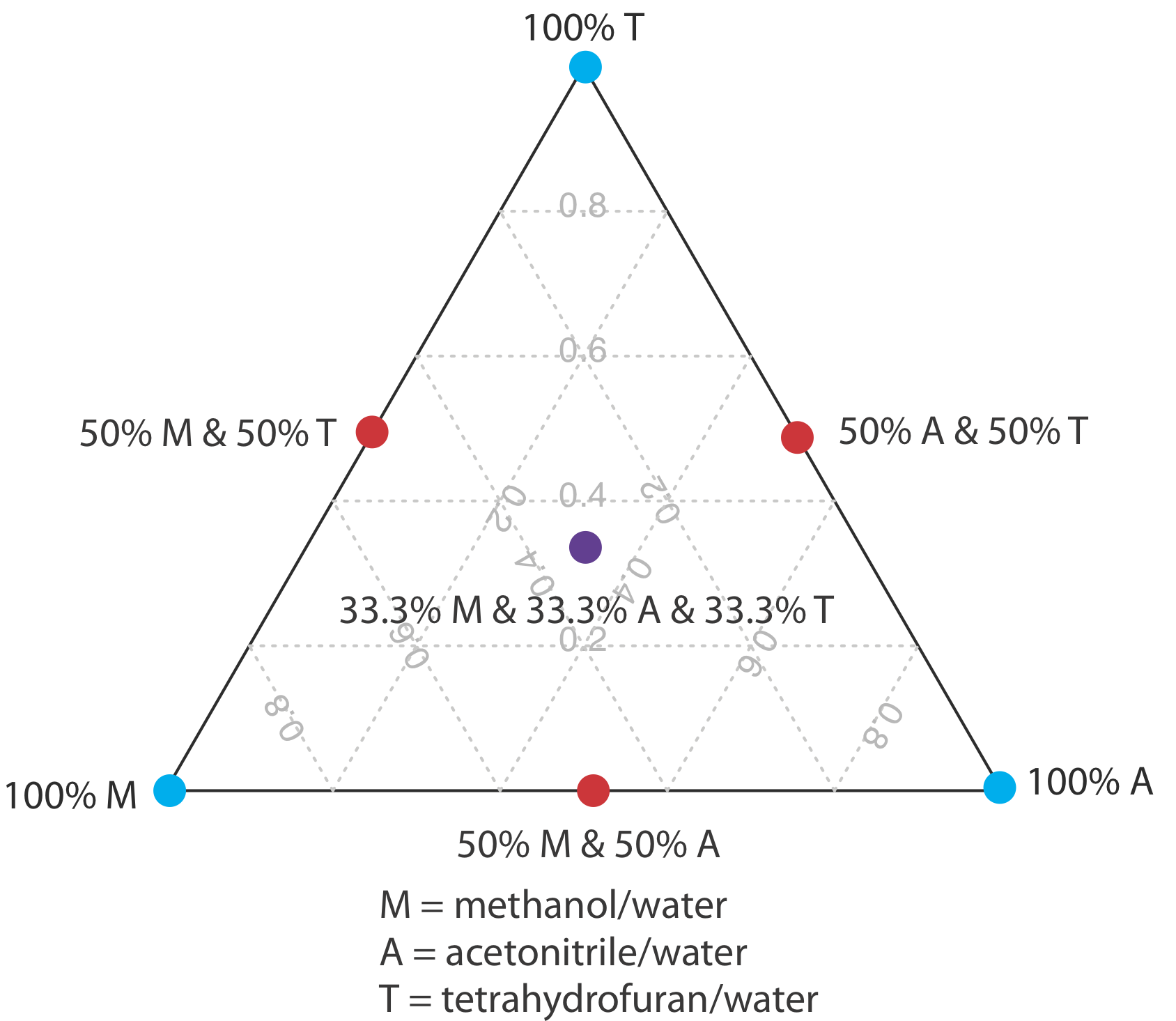
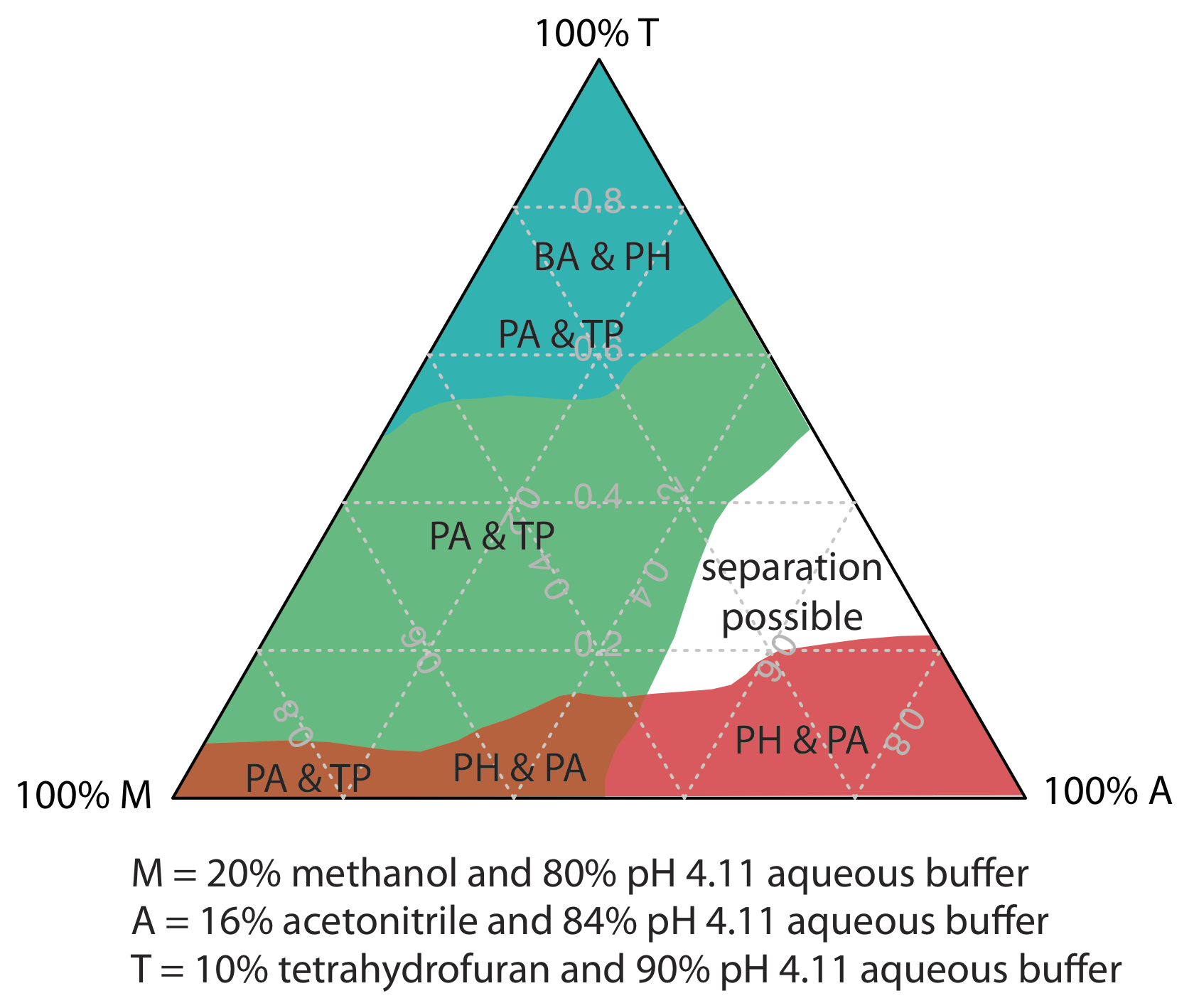
Una estrategia para encontrar la mejor fase móvil es utilizar el triángulo solvente mostrado en la Figura 28.4.2 , lo que nos permite explorar una amplia gama de fases móviles con solo siete experimentos. Comenzamos ajustando la cantidad de acetonitrilo en la fase móvil para producir la mejor separación posible dentro del tiempo de análisis deseado. A continuación, utilizamos la Tabla 28.4.3 para estimar la composición de las fases móviles de metanol/h 2 O y tetrahidrofurano/h 2 O que producirán tiempos de análisis similares. Se preparan cuatro fases móviles adicionales utilizando las fases móviles binarias y ternarias mostradas en la Figura 28.4.2 . Cuando examinamos los cromatogramas de estas siete fases móviles podemos encontrar que una o más proporcionan una separación adecuada, o podemos identificar una región dentro del triángulo solvente donde una separación es factible. La Figura 28.4.3 muestra un mapa de resolución para la separación en fase inversa de ácido benzoico, ácido tereftálico, ácido p-aminbenzoico y ácido p-hidroxibenzoico en una columna C 18 no polar en la que el tiempo máximo de análisis deseado se establece en 6 min [Harvey, D. T.; Byerly, S.; Bowman, A.; Tomlin, J. J. Chem. Educ. 1991, 68, 162—168]. Las áreas en azul, verde y rojo muestran composiciones de fase móvil que no proporcionan resolución basal. El área no sombreada representa composiciones de fase móvil donde es posible una separación.
La elección para comenzar con acetonitrilo es arbitraria; podemos elegir con la misma facilidad comenzar con metanol o tetrahidrofurano.
| %v/v CH 3 OH | % v/v CH 3 CN | %v/v THF |
|---|---|---|
| 0 | 0 | 0 |
| 10 | 6 | 4 |
| 20 | 14 | 10 |
| 30 | 22 | 16 |
| 40 | 32 | 24 |
| 50 | 40 | 30 |
| 6 | 50 | 36 |
| 70 | 60 | 44 |
| 80 | 72 | 52 |
| 90 | 87 | 62 |
| 100 | 99 | 71 |
Elección de una Fase Móvil: Eluciones Isocráticas y Gradientes
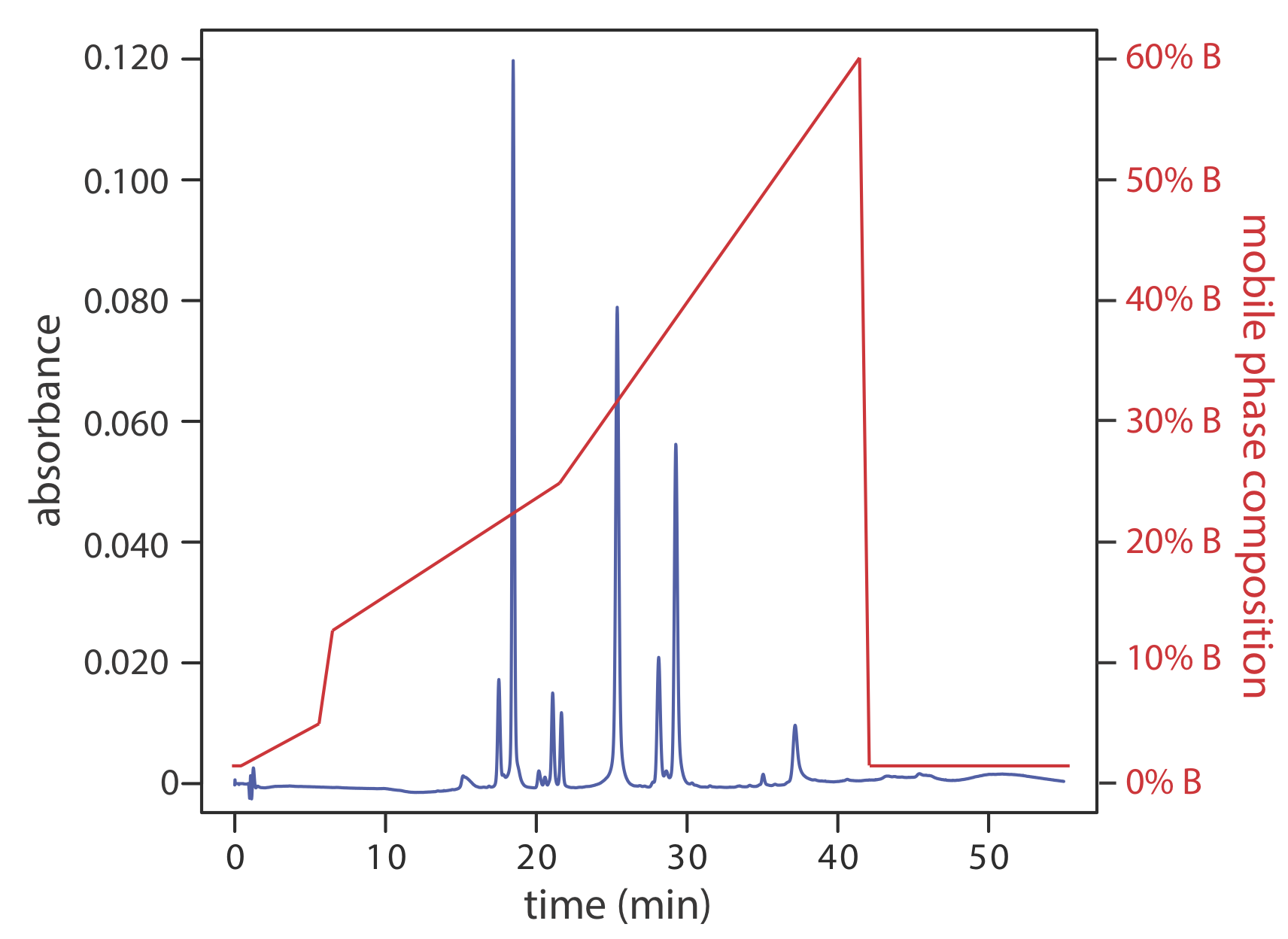
Una separación usando una fase móvil que tiene una composición fija es una elución i socrática. Una dificultad con una elución isocrática es que una fuerza de fase móvil apropiada para resolver solutos de elución temprana puede conducir a tiempos de retención inaceptablemente largos para solutos de elución tardía. La optimización de la fase móvil para solutos de elución tardía, por otro lado, puede proporcionar una separación inadecuada de solutos de elución temprana. Cambiar la composición de la fase móvil a medida que avanza la separación es una solución a este problema. Para una separación de fase inversa utilizamos una fase móvil inicial que es más polar. A medida que avanza la separación, ajustamos la composición de la fase móvil para que se vuelva menos polar (ver Figura 28.4.4 ). Tales separaciones se denominan eluciones de gradiente.
Elegir un detector
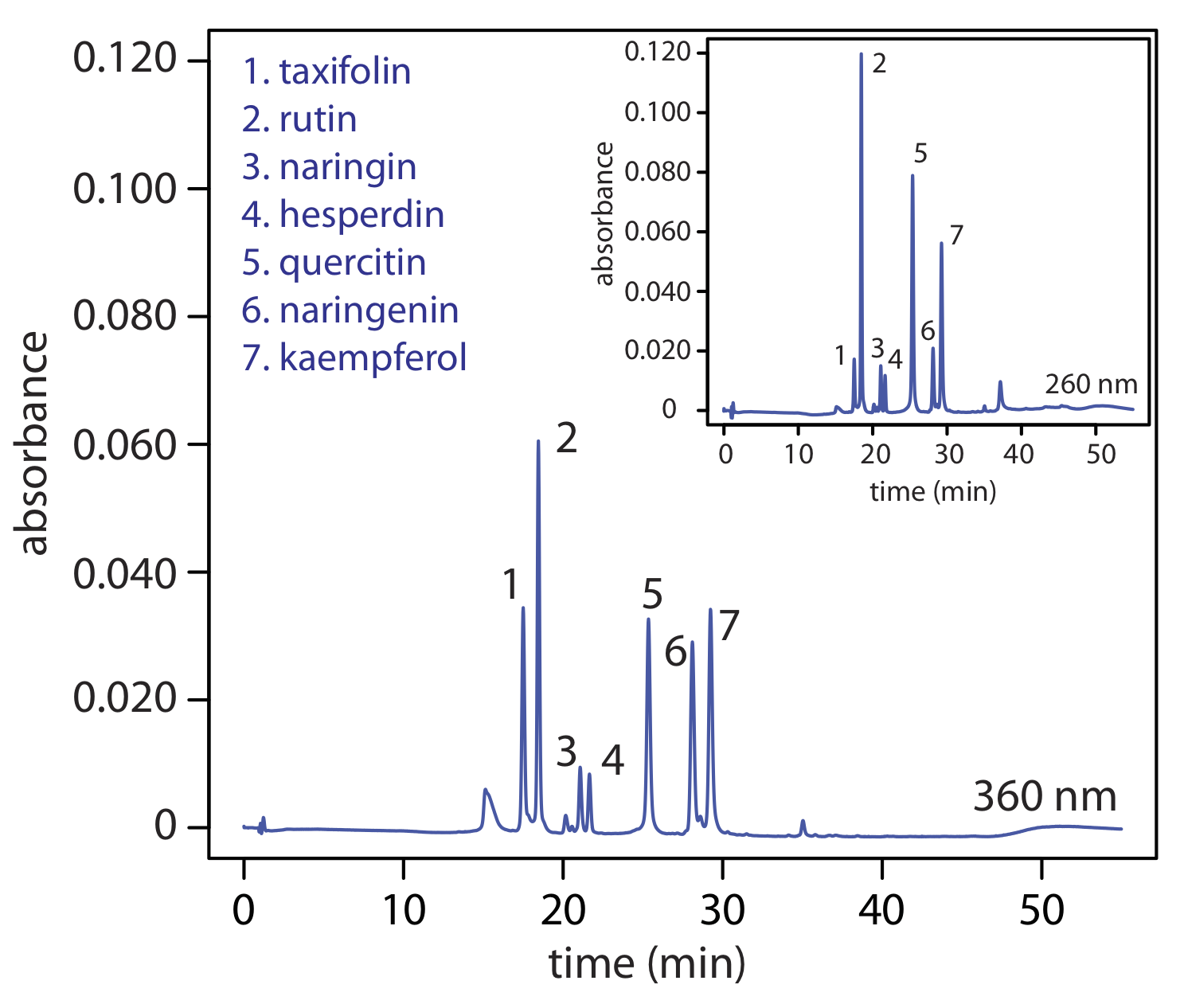
La disponibilidad de diferentes tipos de detectores proporciona otra forma de incorporar selectividad en un análisis. La Figura 28.4.50 , por ejemplo, muestra la separación de fase inversa de una mezcla de flavonoides mediante detección UV/Vis a dos longitudes de onda diferentes. En este caso, una longitud de onda de 260 nm aumenta la sensibilidad del método para la rutina en relación con la de la taxifolina.
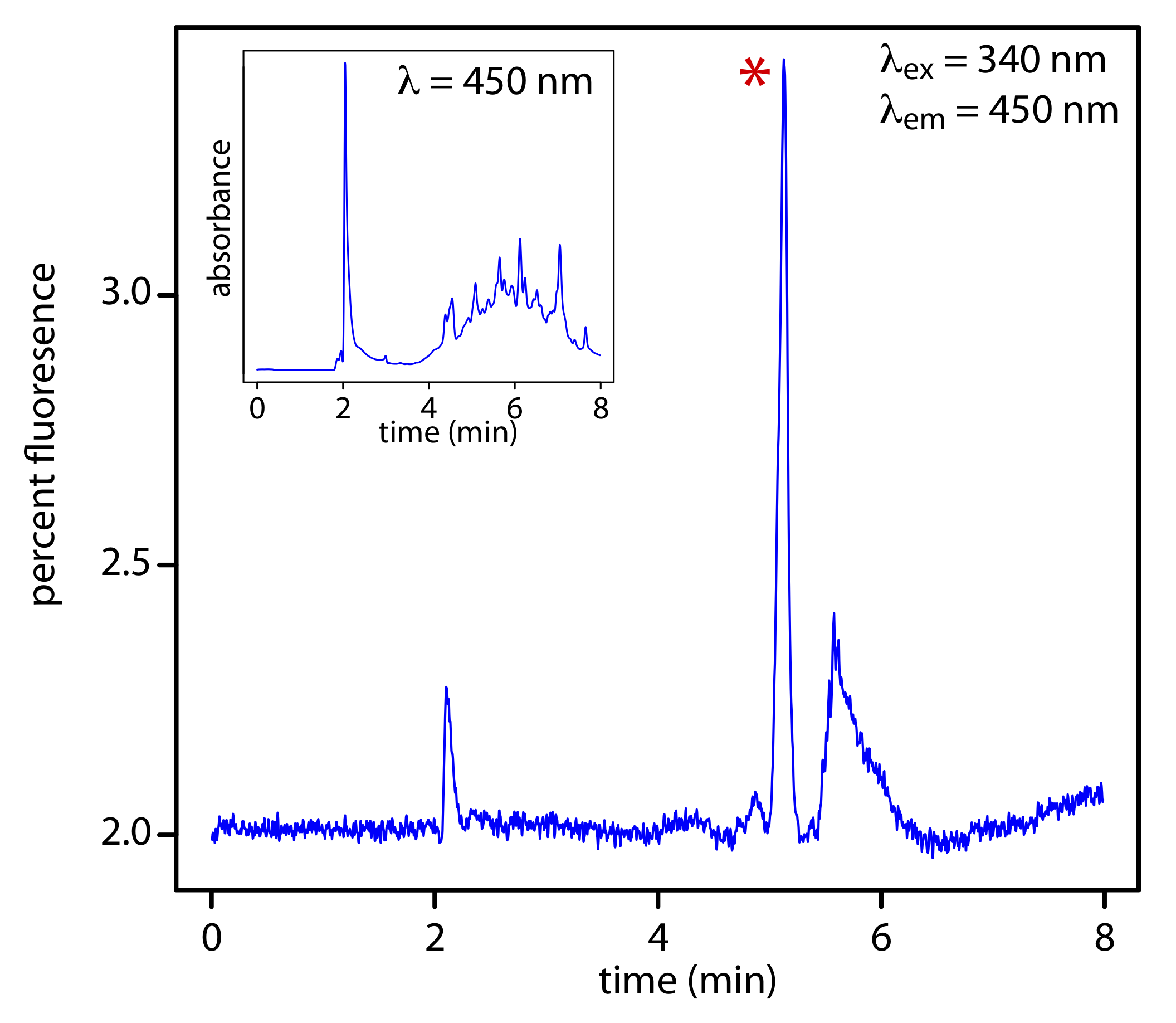
Como se muestra en la Figura 28.4.6 , un detector de fluorescencia proporciona selectividad adicional porque solo unos pocos de los componentes de una muestra son fluorescentes.
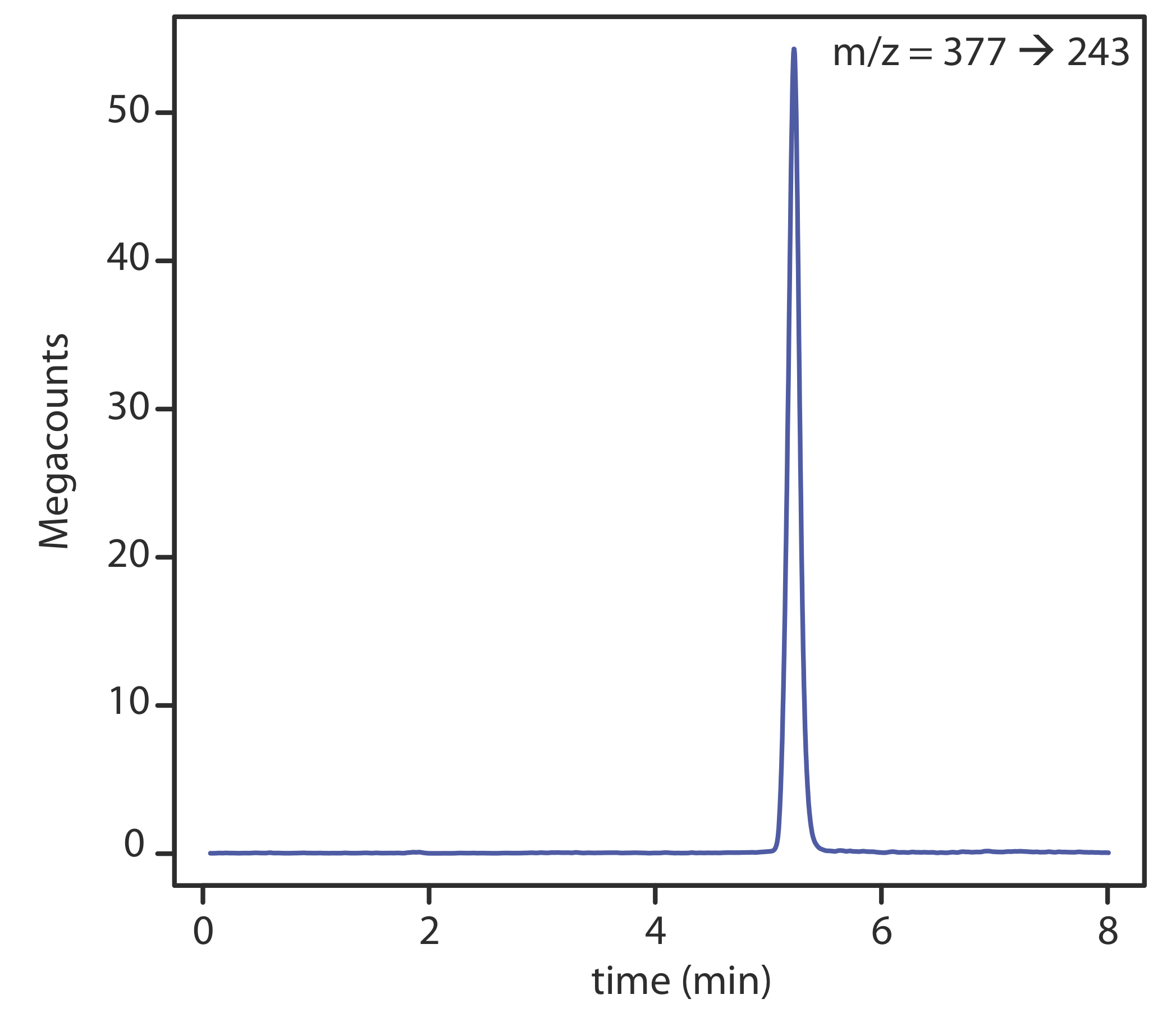
Con un espectrómetro de masas como detector, existen varias opciones para monitorear el cromatograma. El método más común es escanear continuamente todo el espectro de masas y reportar la señal total para todos los iones que llegan al detector durante cada exploración. Este escaneo de iones totales proporciona detección universal para todos los analitos. Como se ve en la Figura 28.4.7 , podemos lograr cierto grado de selectividad monitoreando solo relaciones masa-carga específicas, un proceso llamado monitoreo de iones selectivos.
Aplicaciones cuantitativas de la cromatografía de partición
La cromatografía de partición se utiliza rutinariamente para análisis cualitativos y cuantitativos de muestras ambientales, farmacéuticas, industriales, forenses, clínicas y de productos de consumo.
Preparación de muestras para análisis
Las muestras en forma líquida se inyectan en la HPLC después de una limpieza adecuada para eliminar cualquier material particulado, o después de una extracción adecuada para eliminar los interferentes de la matriz. En la determinación de hidrocarburos poliaromáticos (PAH) en aguas residuales, por ejemplo, una extracción con CH 2 Cl 2 sirve para el doble propósito de concentrar los analitos y aislarlos de los interferentes de la matriz. Las muestras sólidas se disuelven primero en un disolvente adecuado o los analitos de interés se ponen en solución por extracción. Por ejemplo, un análisis de HPLC para los ingredientes activos y los productos de degradación en un comprimido farmacéutico a menudo comienza extrayendo el comprimido en polvo con una porción de fase móvil. Las muestras de gas se recolectan burbujeándolas a través de una trampa que contiene un solvente adecuado. Los isocianatos orgánicos en atmósferas industriales se recolectan burbujeando el aire a través de una solución de 1- (2-metoxifenil) piperazina en tolueno. La reacción entre los isocianatos y la 1- (2-metoxifenil) piperazina los estabiliza frente a la degradación antes del análisis por HPLC y los convierte en una forma química que puede ser monitoreada por absorción UV.
Cálculos cuantitativos
Un análisis por HPLC cuantitativo suele ser más fácil que un análisis de GC cuantitativo porque un bucle de muestra de volumen fijo proporciona una inyección más precisa y precisa. Como resultado, la mayoría de los métodos de HPLC cuantitativa no necesitan un estándar interno y, en su lugar, utilizan estándares externos y una curva de calibración normal.
Un estándar interno es necesario cuando se usa HPLC-MS porque la interfaz entre la HPLC y el espectrómetro de masas no permite una transferencia reproducible del eluyente de la columna a la cámara de ionización de la EM.
La concentración de hidrocarburos aromáticos polinucleares (PAH) en el suelo se determina extrayendo primero los HAP con cloruro de metileno. El extracto se diluye, si es necesario, y los HAP se separan por HPLC usando un detector de UV/Vis o fluorescencia. La calibración se logra usando uno o más estándares externos. En un análisis típico se extrae una muestra de 2.013 g de suelo seco con 20.00 mL de cloruro de metileno. Después de filtrar para eliminar la tierra, se retira una porción de 1.00-mL del extracto y se diluye a 10.00 mL con acetonitrilo. Inyectar 5 μL del extracto diluido en una HPLC da una señal de 0.217 (unidades arbitrarias) para el fluoranteno PAH. Cuando se analizan 5 μL de un estándar de fluoranteno de 20.0-ppm usando las mismas condiciones, se mide una señal de 0.258. Reportan las partes por millón de fluoranteno en el suelo.
Solución
Para un estándar externo de punto único, la relación entre la señal, S, y la concentración, C, de fluoranteno es
\[S = kC \nonumber \]
Sustituyendo en valores la señal y concentración del estándar da el valor de k como
\[k=\frac{S}{C}=\frac{0.258}{20.0 \text{ ppm}}=0.0129 \text{ ppm}^{-1} \nonumber \]
El uso de este valor para k y la señal de HPLC de la muestra da una concentración de fluoranteno de
\[C=\frac{S}{k}=\frac{0.217}{0.0129 \text{ ppm}^{-1}}=16.8 \text{ ppm} \nonumber \]
para la muestra de suelo extraída y diluida. La concentración de fluoranteno en el suelo es
\[\frac{16.8 \text{ g} / \mathrm{mL} \times \frac{10.00 \text{ mL}}{1.00 \text{ mL}} \times 20.00 \text{ mL}}{2.013 \text{ g} \text { sample }}=1670 \text{ ppm} \text { fluoranthene } \nonumber \]
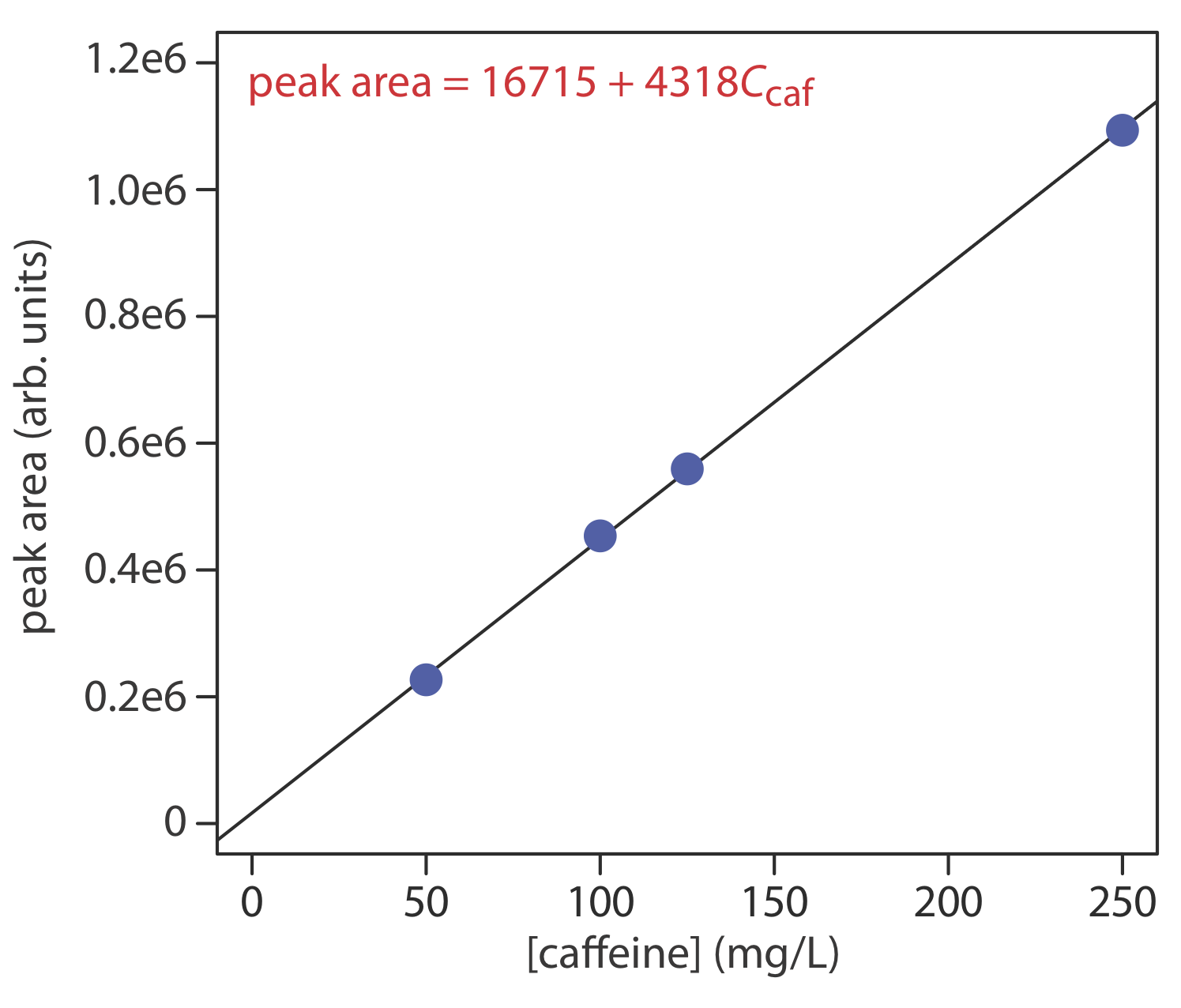
La concentración de cafeína en las bebidas se determina mediante una separación HPLC de fase inversa usando una fase móvil de 20% acetonitrilo y 80% agua, y usando una columna C 8 no polar. Los resultados de una serie de inyecciones de 10-μL de estándares de cafeína se encuentran en la siguiente tabla.
| [cafeína] (mg/L) | área de pico (unidades arb.) |
|---|---|
| 50.0 | 226724 |
| 100.0 | 453762 |
| 125.0 | 559443 |
| 250.0 | 1093637 |
¿Cuál es la concentración de cafeína en una muestra si una inyección de 10 μL da un área pico de 424195? Los datos en este problema provienen de Kusch, P.; Knupp, G. “Determinación Simultánea de Cafeína en Bebidas Cola y Otras Bebidas por HPTLC de Fase Inversa y HPLC de Fase Inversa”, Chem. Educador, 2003, 8, 201—205.
- Contestar
-
La siguiente figura muestra la curva de calibración y la ecuación de calibración para el conjunto de estándares externos. Al sustituir el área del pico de la muestra en la ecuación de calibración se obtiene la concentración de cafeína en la muestra como 94.4 mg/L.