
28.3: Instrumentos para Cromatografía Líquida
- Page ID
- 79048

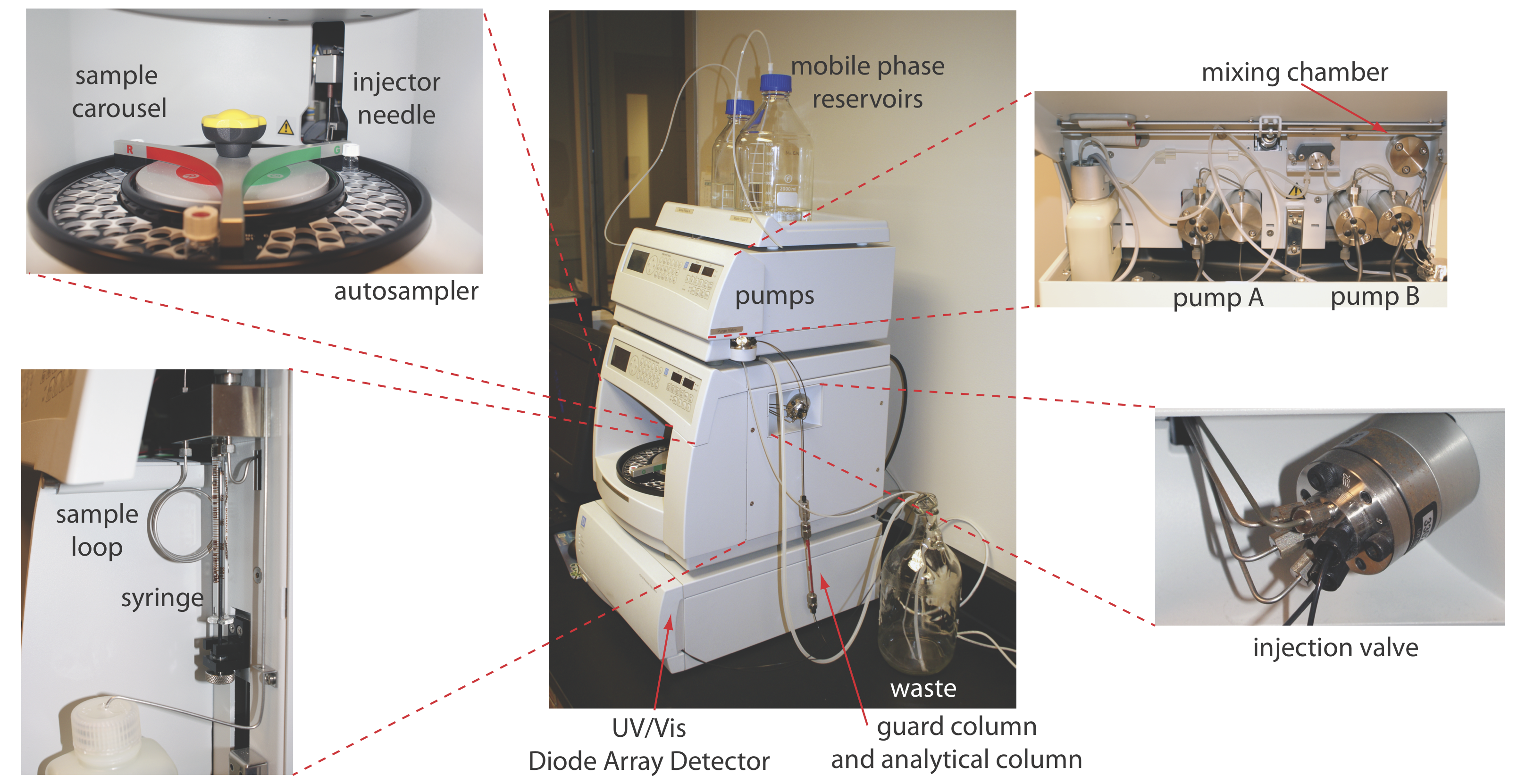
En cromatografía líquida de alta resolución (HPLC) inyectamos la muestra, que está en forma de solución, en una fase móvil líquida. La fase móvil transporta la muestra a través de una columna empaquetada o capilar que separa los componentes de la muestra en función de su capacidad de partición entre la fase móvil y la fase estacionaria. La Figura 28.3.1 muestra un ejemplo de un instrumento típico de HPLC, que tiene varios componentes clave: depósitos que almacenan la fase móvil; una bomba para empujar la fase móvil a través del sistema; un inyector para introducir la muestra; una columna para separar la muestra en sus partes componentes; y un detector para monitorear el eluyente a medida que sale de la columna. Consideremos cada uno de estos componentes.
Columnas de HPLC
Una HPLC normalmente incluye dos columnas: una columna analítica, que es responsable de la separación, y una columna de protección que se coloca antes de la columna analítica para protegerla de la contaminación.
Columnas Analíticas
El tipo más común de columna de HPLC es un tubo de acero inoxidable con un diámetro interno entre 2.1 mm y 4.6 mm y una longitud entre 30 mm y 300 mm (Figura 28.3.2 ). La columna está empaquetada con partículas porosas de sílice de 3—10 µm con forma irregular o esférica. Las eficiencias típicas de la columna son 40 000—60 000 placas teóricas/m. Una columna de 25 cm con 50 000 placas/m tiene 12 500 placas teóricas.

Las columnas capilares utilizan menos disolvente y, debido a que la muestra se diluye en menor medida, producen señales más grandes en el detector. Estas columnas están hechas de capilares de sílice fundida con diámetros internos de 44—200 μm y longitudes de 50—250 mm. Se han preparado columnas capilares empaquetadas con partículas de 3—5 μm con eficiencias de columna de hasta 250 000 placas teóricas [Novotony, M. Science, 1989, 246, 51—57].
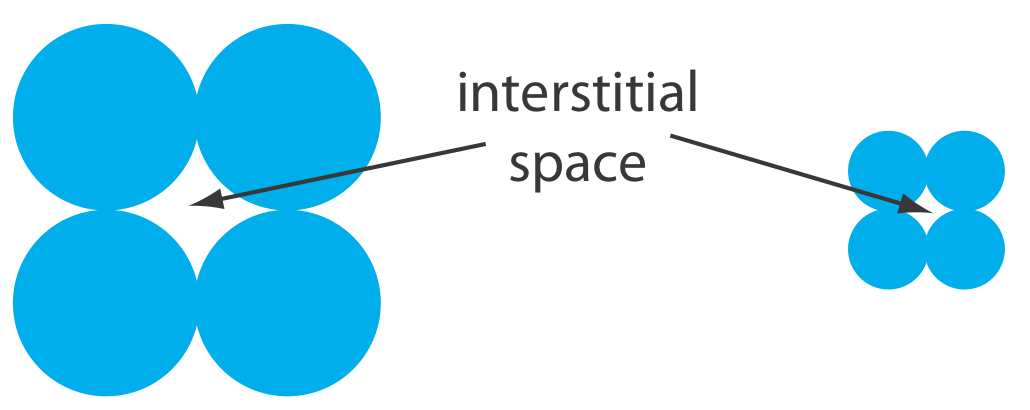
Una limitación para una columna capilar empaquetada es la contrapresión que se desarrolla al bombear la fase móvil a través de los pequeños espacios intersticiales entre el material de empaquetamiento de tamaño micrométrico particulado (Figura 28.3.3 ). Debido a que los tubos y los accesorios que llevan la fase móvil tienen límites de presión, una contrapresión más alta requiere un caudal más bajo y un tiempo de análisis más largo. Las columnas monolíticas, en las que el soporte sólido es una sola varilla porosa, ofrecen eficiencias de columna equivalentes a una columna capilar empaquetada al tiempo que permiten caudales más rápidos. Una columna monolítica, que generalmente es similar en tamaño a una columna empaquetada convencional, aunque también hay disponibles columnas capilares más pequeñas, se prepara formando la varilla monolítica en un molde y cubriéndola con tubos de PTFE o una resina polimérica. Las varillas monolíticas hechas de un polímero de gel de sílice-gel suelen tener macroporos con diámetros de aproximadamente 2 μm y mesoporos—poros dentro de los macroporos—con diámetros de aproximadamente 13 nm [Cabrera, K. Chromatography Online, 1 de abril de 2008].
Columnas de Guardia
Dos problemas tienden a acortar la vida útil de una columna analítica. Primero, los solutos que se unen irreversiblemente a la fase estacionaria degradan el rendimiento de la columna al disminuir la cantidad de fase estacionaria disponible para efectuar una separación. Segundo, el material particulado inyectado con la muestra puede obstruir la columna analítica. Para minimizar estos problemas colocamos una columna de guarda antes de la columna analítica. Una columna Guard generalmente contiene el mismo material de relleno particulado y fase estacionaria que la columna analítica, pero es significativamente más corta y menos cara, una longitud de 7.5 mm y un costo de una décima parte del de la columna analítica correspondiente es típica. Debido a que están destinados a ser sacrificiales, las columnas de guardia se reemplazan regularmente. Si observa de cerca la Figura 28.3.1 , verá la pequeña columna de guarda justo encima de la columna analítica.
Plomería HPLC
En un cromatógrafo de gases la presión de un cilindro de gas comprimido es suficiente para empujar la fase móvil a través de la columna. Empujar una fase móvil líquida a través de una columna, sin embargo, requiere mucho más esfuerzo, generando presiones superiores a varios cientos de atmósferas. En esta sección consideramos la plomería básica necesaria para mover la fase móvil a través de la columna e inyectar la muestra a la fase móvil.
Mover la fase móvil
Una HPLC típica incluye entre 1 y 4 depósitos para almacenar disolventes de fase móvil. El instrumento en la Figura 28.3.1 , por ejemplo, tiene dos depósitos de fase móvil que se utilizan para una elución isocrática o una elución en gradiente mediante la extracción de disolventes de uno o ambos depósitos.
Antes de usar un disolvente de fase móvil debemos eliminar los gases disueltos, como N 2 y O 2, y pequeñas partículas, como el polvo. Debido a que hay una gran caída de presión a través de la columna, la presión en la entrada de la columna es de varios cientos de atmósferas, pero es la presión atmosférica a la salida de la columna, los gases disueltos en la fase móvil se liberan como burbujas de gas que pueden interferir con la respuesta del detector. La desgasificación se realiza de varias maneras, pero las más comunes son el uso de una bomba de vacío o el burbujeo con un gas inerte, como el He, que tiene una baja solubilidad en la fase móvil. Los materiales particulados, que pueden obstruir el tubo o columna de HPLC, se eliminan filtrando los disolventes.
El burbujeo de un gas inerte a través de la fase móvil libera gases volátiles disueltos. Este proceso se llama sparging.
Los disolventes de fase móvil se extraen de sus depósitos por la acción de una o más bombas. La Figura 28.3.4 muestra una vista en primer plano de las bombas para el instrumento en la Figura 28.3.1 . La bomba de trabajo y la bomba equilibradora tienen cada una un pistón cuyo movimiento de ida y vuelta mantiene un caudal constante de hasta varios ml/min y proporciona la alta presión de salida necesaria para empujar la fase móvil a través de la columna cromatográfica. En este instrumento en particular, cada bomba envía su fase móvil a una cámara de mezcla donde se combinan para formar la fase móvil final. La velocidad relativa de las dos bombas determina la composición final de la fase móvil.
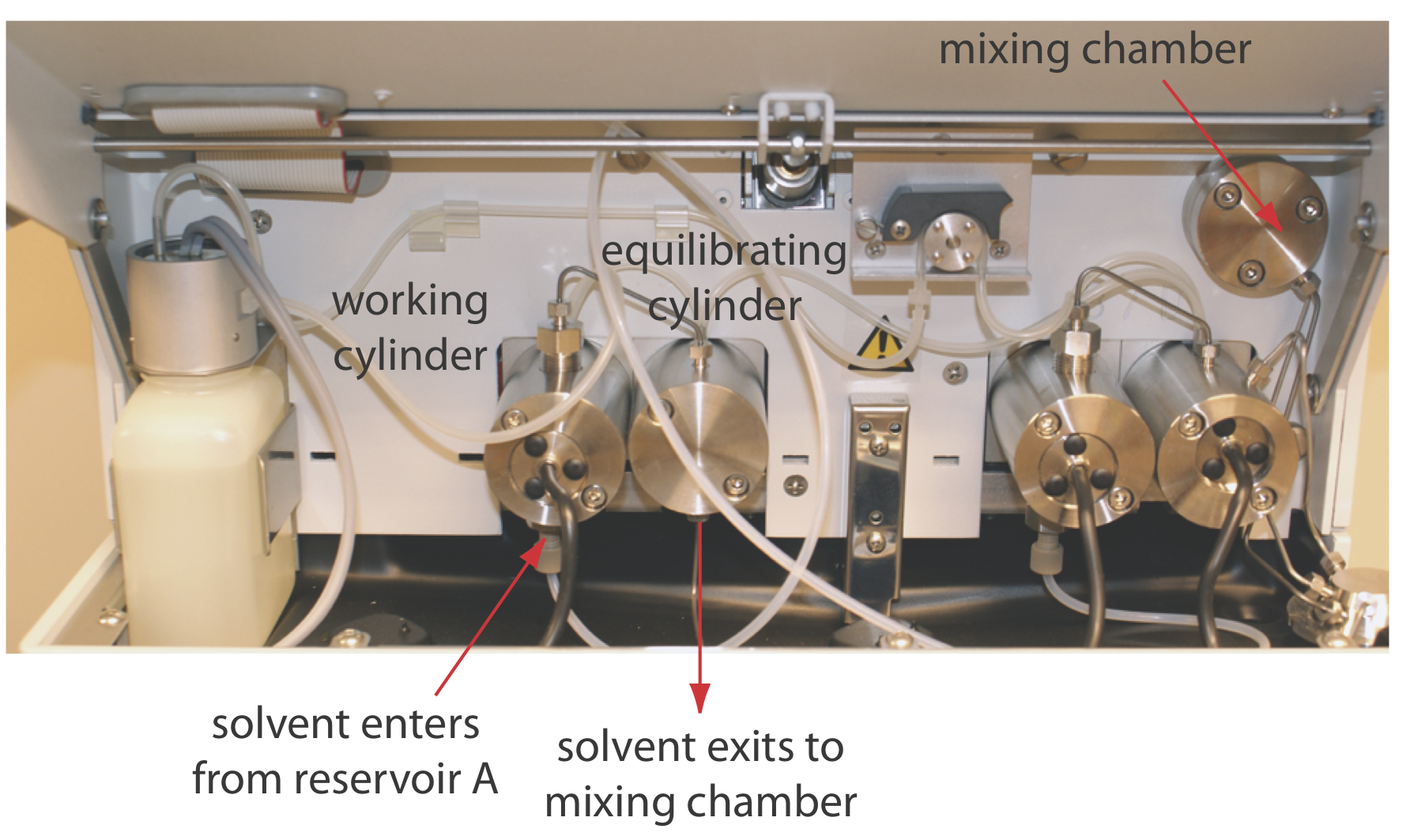
El movimiento de ida y vuelta de una bomba de vaivén crea un flujo pulsado que aporta ruido al cromatograma. Para minimizar estos pulsos, cada bomba de la Figura 28.3.4 tiene dos cilindros. Durante el movimiento delantero del cilindro de trabajo, llena el cilindro equilibrador y establece el flujo a través de la columna. Cuando el cilindro de trabajo está en su carrera inversa, el flujo es mantenido por el pistón en el cilindro de equilibrio. El resultado es un flujo libre de pulsos.
Hay otras formas de controlar la composición y el caudal de la fase móvil. Por ejemplo, en lugar de las dos bombas de la Figura 28.3.4 , podemos colocar una válvula dosificadora de disolvente antes de una sola bomba. El valor de dosificación de solvente conecta dos o más depósitos de solvente a la bomba y determina cuánto de cada solvente se extrae durante cada uno de los ciclos de la bomba. Otro enfoque para eliminar un flujo pulsado es incluir un amortiguador de pulsos entre la bomba y la columna. Un amortiguador de pulsos es una cámara llena de un fluido fácilmente comprimido y un diafragma flexible. Durante la carrera hacia adelante del pistón, el fluido en el amortiguador de impulsos se comprime. Cuando el pistón se retira para rellenar la bomba, la presión del fluido de expansión en el amortiguador de pulsos mantiene el caudal.
Inyectar la muestra
La presión de operación dentro de una HPLC es suficientemente alta como para que no podamos inyectar la muestra en la fase móvil insertando una jeringa a través de un septo, como es posible en la cromatografía de gases. En su lugar, inyectamos la muestra usando un inyector de bucle, cuyo diagrama se muestra en la Figura 28.3.5 . En la posición de carga, un bucle de muestra, que está disponible en una variedad de tamaños que van desde 0.5 μL hasta 5 ml, se aísla de la fase móvil y se abre a la atmósfera. El asa de muestra se llena usando una jeringa con una capacidad varias veces mayor que la del asa de muestra, con exceso de muestra saliendo por la línea de desechos. Después de cargar la muestra, el inyector se gira a la posición de inyección, que redirige la fase móvil a través del bucle de muestra y hacia la columna.
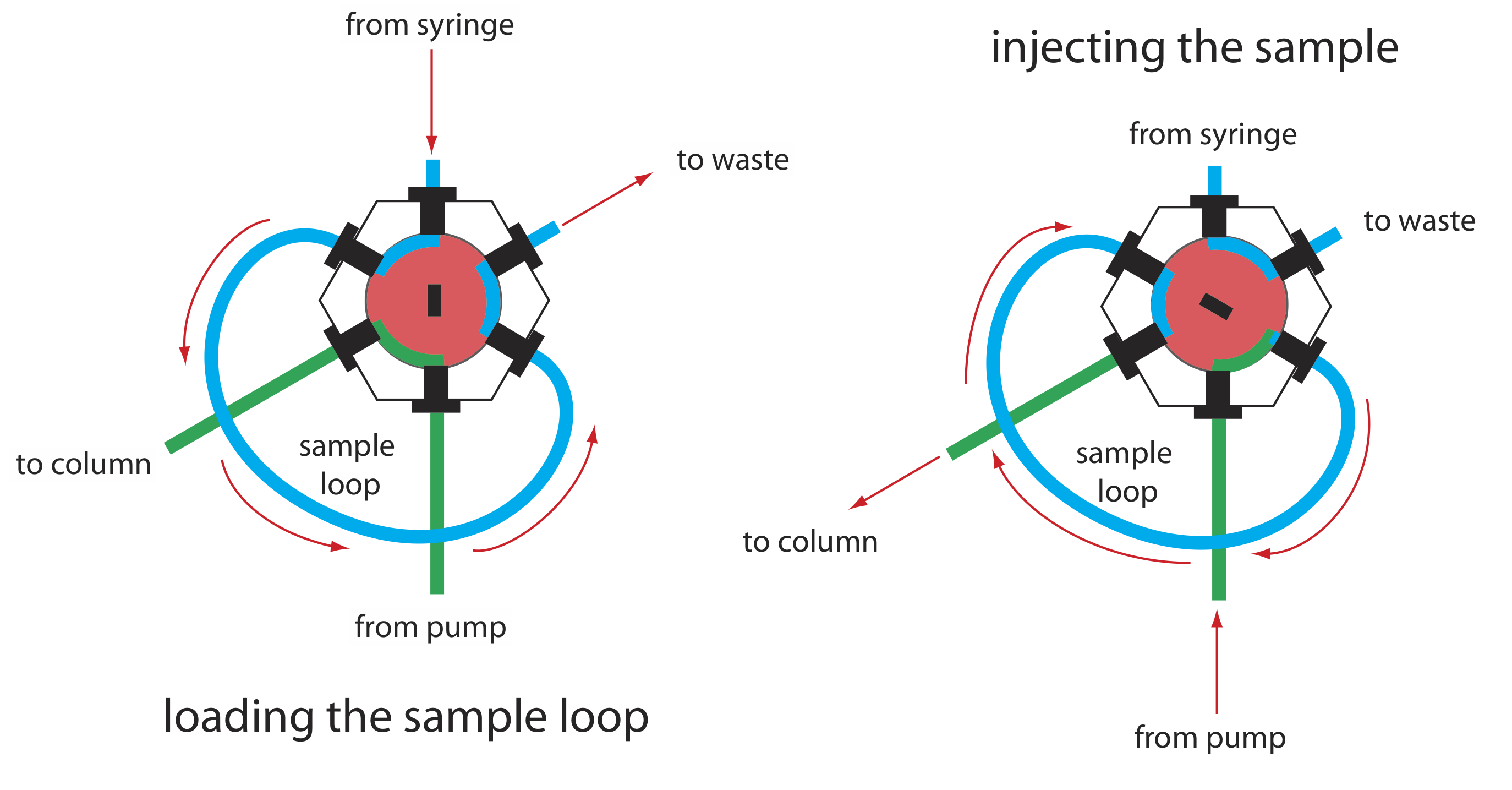
El instrumento en la Figura 28.3.1 utiliza un muestreador automático para inyectar muestras. En lugar de usar una jeringa para empujar la muestra hacia el bucle de muestra, la jeringa extrae la muestra hacia el bucle de muestra.
Detectores para HPLC
Se han utilizado muchos tipos diferentes de detectores para monitorear las separaciones por HPLC, la mayoría de los cuales utilizan espectroscopía o electroquímica para generar una señal medible.
Detectores espectroscópicos
Los detectores de HPLC más populares aprovechan el espectro de absorción UV/Vis de un analito. Estos detectores van desde diseños simples, en los que la longitud de onda analítica se selecciona utilizando filtros apropiados, hasta un espectrofotómetro modificado en el que el compartimento de muestra incluye una celda de flujo. La Figura 28.3.6 muestra el diseño de una celda de flujo típica cuando se usa un espectrómetro de matriz de diodos como detector. La celda de flujo tiene un volumen de 1—10 μL y una longitud de trayectoria de 0.2—1 cm.
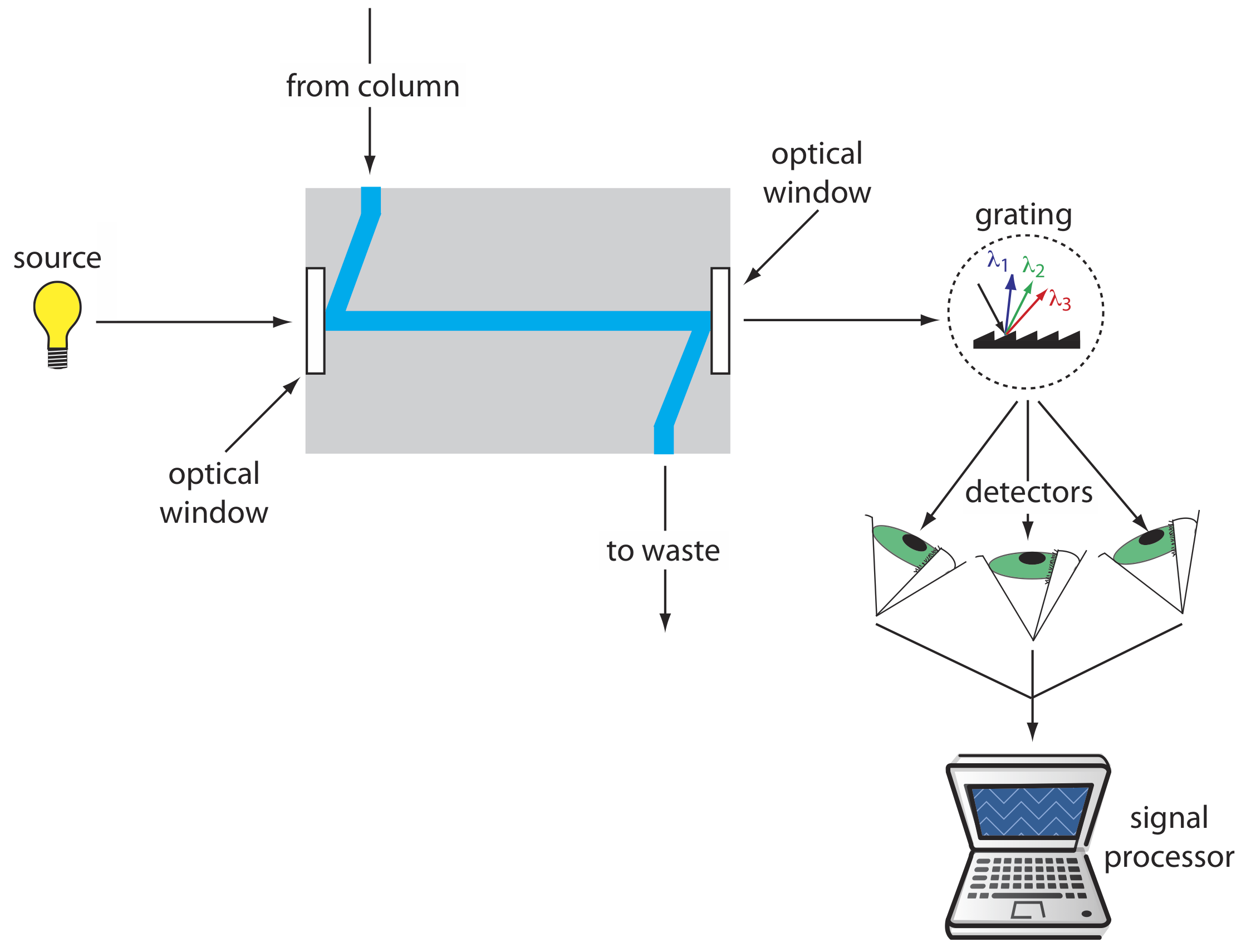
Cuando se usa un detector UV/Vis el cromatograma resultante es una gráfica de absorbancia en función del tiempo de elución. Si el detector es un espectrómetro de matriz de diodos, entonces también podemos mostrar el resultado como un cromatograma tridimensional que muestra la absorbancia en función de la longitud de onda y el tiempo de elución. Una limitación al uso de la absorbancia es que la fase móvil no puede absorber en las longitudes de onda que deseamos monitorear. Los detectores de absorbancia proporcionan límites de detección de tan solo 100 pg-1 ng de analito inyectado. Si un analito es fluorescente, podemos colocar la celda de flujo en un espectrofluorímetro. Los límites de detección son tan pequeños como 1—10 pg de analito inyectado.
Detectores Electroquímicos
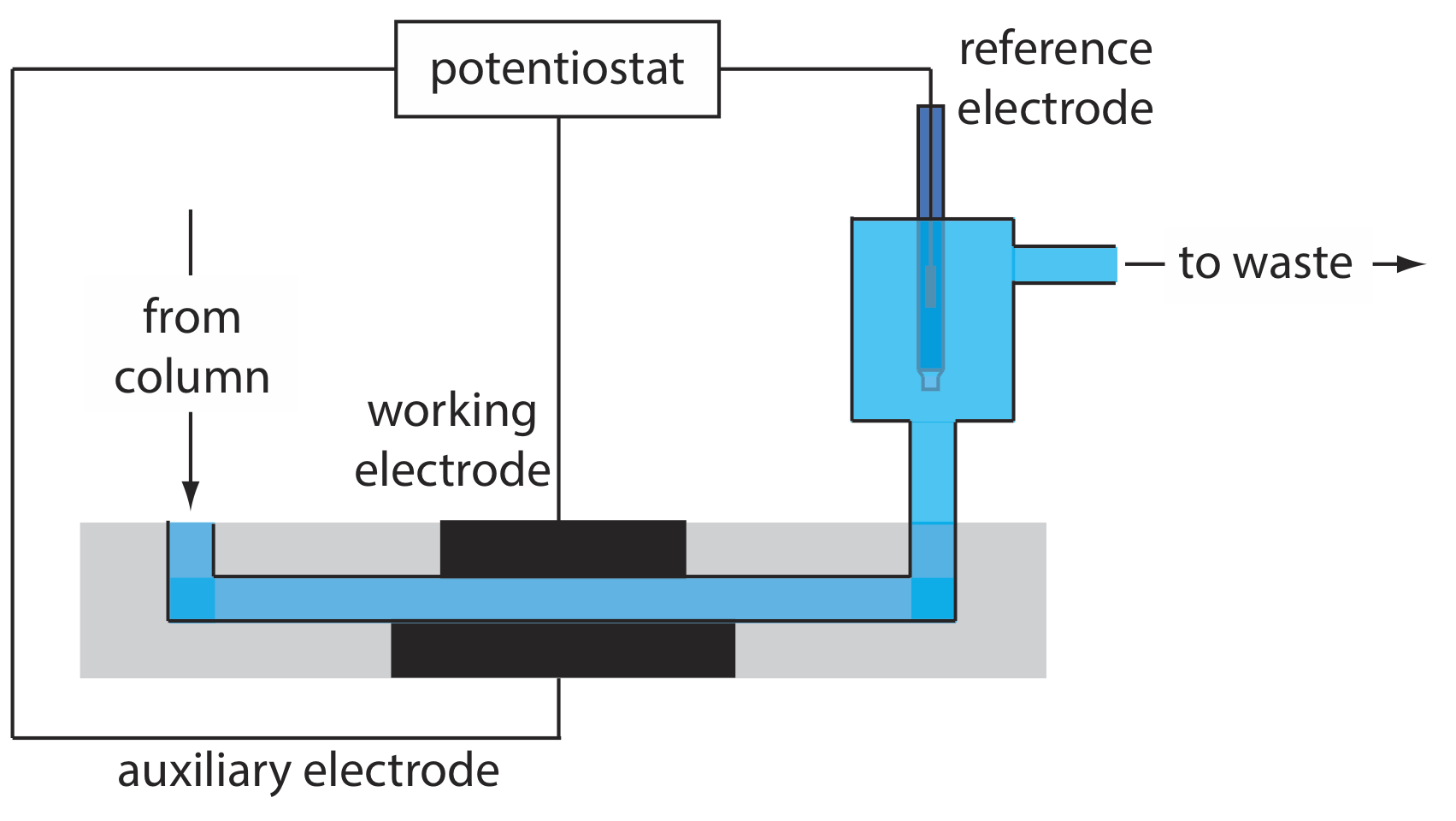
Otro grupo común de detectores de HPLC son aquellos basados en mediciones electroquímicas como amperometría, voltamperometría, culombimetría y conductividad. La figura 28.3.7 , por ejemplo, muestra una celda de flujo amperométrico. El efluente de la columna pasa sobre el electrodo de trabajo, mantenido a un potencial constante en relación con un electrodo de referencia aguas abajo, que oxida o reduce completamente los analitos. La corriente que fluye entre el electrodo de trabajo y el electrodo auxiliar sirve como señal analítica. Los límites de detección para la detección electroquímica amperométrica son de 10 pg-1 ng de analito inyectado.
Otros Detectores
Varios otros detectores han sido utilizados en HPLC. Medir un cambio en el índice de refracción de la fase móvil es análogo a monitorear la conductividad térmica de la fase móvil en cromatografía de gases. Un detector de índice de refracción es casi universal, respondiendo a casi todos los compuestos, pero tiene un límite de detección relativamente pobre de 0.1—1 μg de analito inyectado. Una limitación adicional de un detector de índice de refracción es que no puede usarse para una elución en gradiente a menos que los componentes de la fase móvil tengan índices de refracción idénticos.
Otro detector útil es un espectrómetro de masas. La Figura 28.3.8 muestra un diagrama de bloques de un instrumento HPLC—MS típico. El efluente de la columna ingresa a la fuente de iones del espectrómetro de masas utilizando una interfaz que elimina la mayor parte de la fase móvil, una necesidad esencial debido a la incompatibilidad entre la fase móvil líquida y el ambiente de alto vacío del espectrómetro de masas. En la cámara de ionización las moléculas restantes, una mezcla de los componentes de la fase móvil y los solutos, experimentan ionización y fragmentación. El analizador de masas del espectrómetro de masas separa los iones por su relación masa/carga (m/z). Un detector cuenta los iones y muestra el espectro de masas.
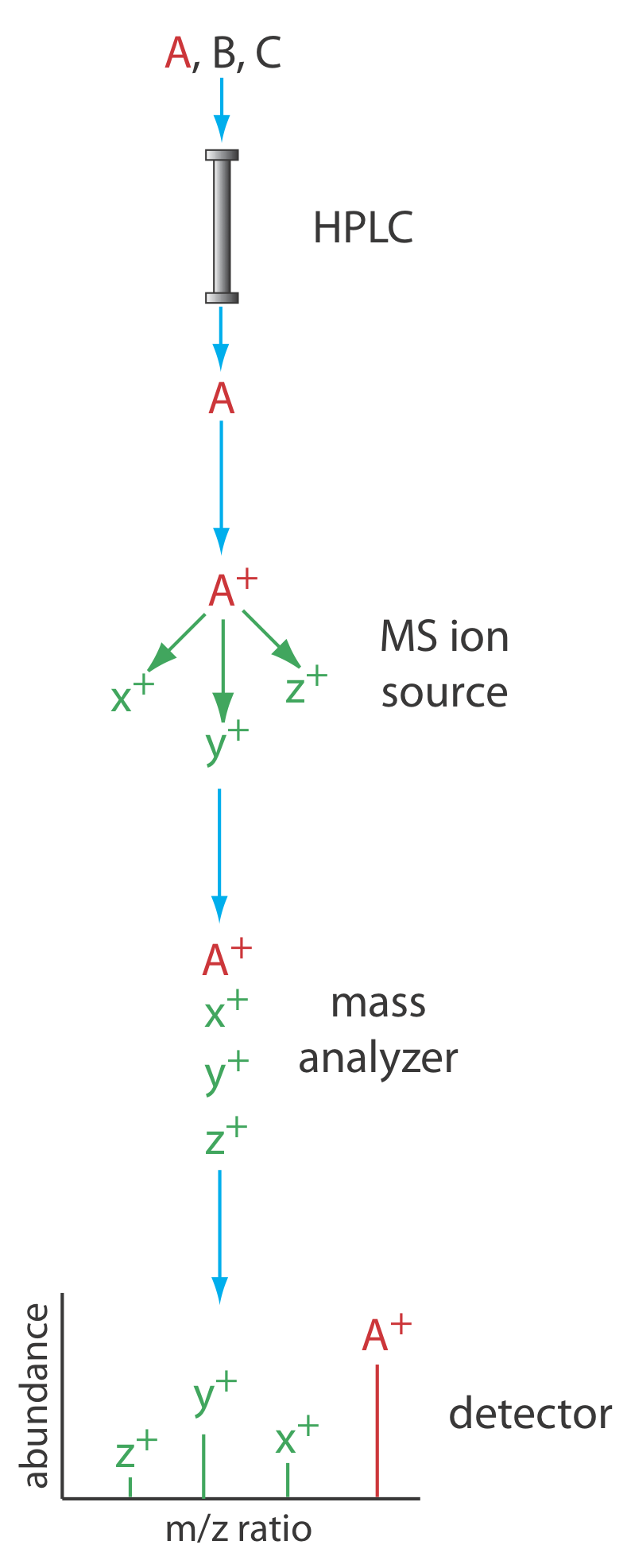
Existen varias opciones para monitorear el cromatograma cuando se usa un espectrómetro de masas como detector. El método más común es escanear continuamente todo el espectro de masas y reportar la señal total para todos los iones que llegan al detector durante cada exploración. Este escaneo de iones totales proporciona detección universal para todos los analitos. Podemos lograr cierto grado de selectividad monitoreando solo relaciones específicas de masa a carga, un proceso llamado monitoreo de iones selectivos. Las ventajas de usar un espectrómetro de masas en HPLC son las mismas que para la cromatografía de gases. Los límites de detección son muy buenos, típicamente 0.1—1 ng de analito inyectado, con valores tan bajos como 1—10 pg para algunas muestras. Además, un espectrómetro de masas proporciona información cualitativa y estructural que puede ayudar a identificar los analitos. La interfaz entre la HPLC y el espectrómetro de masas es técnicamente más difícil que en un GC-MS debido a la incompatibilidad de una fase móvil líquida con el requerimiento de alto vacío del espectrómetro de masas.