1.16: Una Evaluación de Precisión del Refinamiento del Trastorno Metálico Posicional Cristalográfico en Soluciones Sólidas Moleculares
- Page ID
- 71283

Introducción
El trastorno posicional cristalográfico es evidente cuando una posición en la red está ocupada por dos o más átomos; cuyo promedio constituye la composición volumétrica del cristal. Si un átomo en particular ocupa una determinada posición en una celda unitaria y otro átomo ocupa la misma posición en otras celdas unitarias, la densidad electrónica resultante será un promedio ponderal de la situación en todas las celdas unitarias a lo largo del cristal. Dado que el experimento de difracción involucra el promedio de un número muy grande de celdas unitarias (ca. 1018 en un cristal utilizado para el análisis de difracción de rayos X monocristalino), los desplazamientos estáticos menores de los átomos simulan estrechamente los efectos de las vibraciones sobre la potencia de dispersión del átomo “promedio”. Desafortunadamente, la determinación del átomo “promedio” en un cristal puede complicarse si se encuentra un trastorno posicional.
Los trastornos cristalinos que involucran grupos como CO, CN y Cl han sido documentados para crear problemas en la asignación de la estructura correcta a través de procedimientos de refinamiento. Si bien se han hecho intentos de correlacionar los parámetros de la red cristalográfica con la composición química a granel de la solución a partir de la cual se cultivó el monocristal, ha habido poco esfuerzo para correlacionar la ocupación del sitio cristalográfico con la composición química del cristal a partir del cual el monocristal Se obtuvieron datos de difracción. Se trata de dos temas muy diferentes que deben considerarse a la hora de resolver una estructura cristalina con trastorno de ocupación del sitio.
¿Cuál es la relación de un solo cristal con el material a granel?
¿El refinamiento de un factor de ocupación de sitio realmente da un valor realista para% de ocupación cuando se compara con la composición% “real” para ese cristal único en particular?
Lo siguiente representa una descripción de una serie de métodos para el refinamiento de un trastorno de ocupación del sitio entre dos átomos (por ejemplo, dos átomos metálicos dentro de una mezcla de compuestos isoestructurales).
Métodos para la determinación por difracción de rayos X del trastorno posicional en soluciones sólidas moleculares
Un átomo en una estructura se define por varios parámetros: el tipo de átomo, las coordenadas posicionales (x, y, z), el factor de ocupación (cuántos “átomos” hay en esa posición) y los parámetros de desplazamiento atómico (a menudo llamados parámetros de temperatura o térmicos). Este último puede considerarse como una “imagen” del volumen ocupado por el átomo sobre todas las celdas unitarias, y puede ser isotrópico (1 parámetro que define un volumen esférico) o anisotrópico (6 parámetros que definen un volumen elipsoidal). Para un átomo “normal”, el factor de ocupación se fija como igual a uno, y las posiciones y parámetros de desplazamiento se “refinan” utilizando métodos de mínimos cuadrados a valores en los que se obtiene la mejor concordancia con los datos observados. En cristales con desorden de sitio, una posición está ocupada por diferentes átomos en diferentes celdas unitarias. Este refinamiento requiere un enfoque más complicado. Se pueden utilizar dos métodos amplios: o bien se define un nuevo tipo de átomo que es la combinación apropiada de los diferentes átomos, o se utilizan los mismos parámetros posicionales para diferentes átomos en el modelo, cada uno de los cuales tiene valores de ocupación menores a uno, y para los cuales la suma se limita al total de uno. En ambos enfoques se requieren las ocupaciones relativas de los dos átomos. Para el primer acercamiento, se tienen que definir estas ocupaciones. Para el segundo, se puede refinar el valor. Sin embargo, existe una relación entre el parámetro térmico y el valor de ocupación por lo que se debe tener cuidado al hacer esto. Estos temas pueden ser abordados de varias maneras.
Método 1
La suposición más simple es que el cristal a partir del cual se determina la estructura de rayos X representa la muestra a granel que se cristalizó. Con este valor, o bien se puede generar un nuevo tipo de átomo que sea la combinación apropiada de la composición porcentual de tipo 1 (M) de átomo medido y tipo 2 (M') o se pueden ingresar dos átomos diferentes con el factor de ocupación establecido para reflejar la composición porcentual del material a granel. En cualquier caso, se puede permitir que los parámetros térmicos se refinen como de costumbre.
Método 2
Los valores de ocupación para dos átomos (M y M') se refinan (de tal manera que su suma fue igual a 1), mientras que los dos átomos están obligados a tener los mismos parámetros de desplazamiento.
Método 3
Los valores de ocupación (tal que su suma fue igual a 1) y los parámetros de desplazamiento se refinan independientemente para los dos átomos.
Método 4
Una vez obtenidos los mejores valores de ocupación utilizando cualquiera de los Métodos 2 o 3, estos valores se fijaron y se permitió que los parámetros de desplazamiento se refinaran libremente.
Un sistema modelo
Los complejos de metal β -dicetonato (Figura\(\PageIndex{1}\)) para metales en el mismo estado de oxidación son isoestructurales y a menudo isomorfos. Por lo tanto, los cristales obtenidos de la co-cristalización de dos o más complejos de metal β - dicetonato [por ejemplo, Al (acac) 3 y Cr (acac) 3] pueden considerarse como un híbrido de los precursores; es decir, la posición del metal en la red cristalina puede definirse como que tiene el metal promedio composición.
Se pueden preparar una serie de soluciones sólidas de Al (acac) 3 y Cr (acac) 3 para su estudio por difracción de rayos X, mediante la cristalización a partir de soluciones de acetona de mezclas específicas de Al (acac) 3 y Cr (acac) 3 (Cuadro\(\PageIndex{1}\), Columna 1). Los derivados puros y la solución sólida, Al1-xCRx (acac) 3, cristalizan en el grupo espacial monoclínico P21/c con Z = 4.
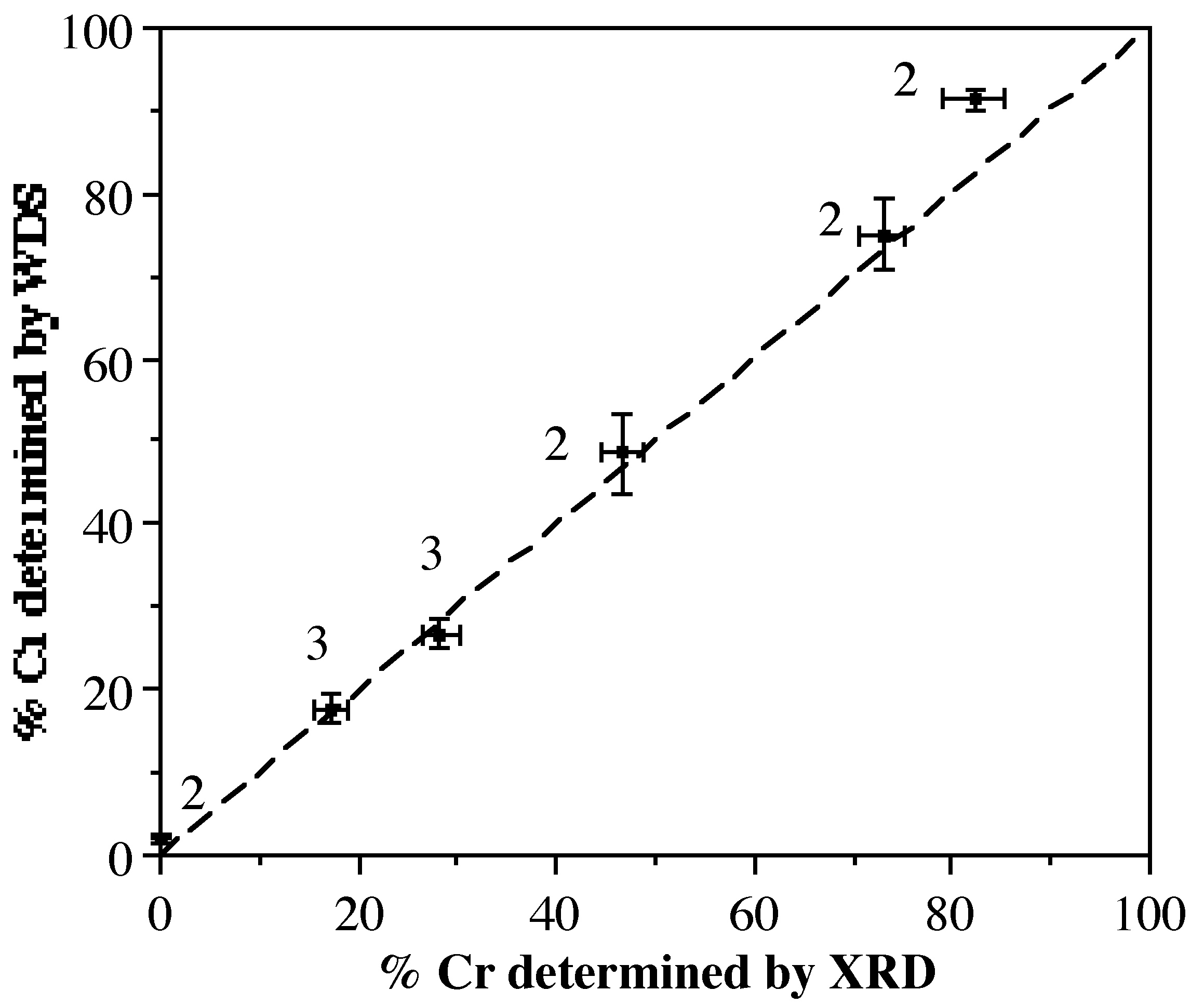
| Composición de la solución (% Cr) | Composición WDS de Cristal Único (% Cr) | Composición refinada de difracción de rayos X (% Cr) |
| 13 | 1.9 ± 0.2 | 0 a |
| 2 | 2.1 ± 0.3 | 0 a |
| 20 | 17.8 ± 1.6 | 17.3 ± 1.8 |
| 26 | 26.7 ± 1.7 | 28.3 ± 1.9 |
| 18 | 48.5 ± 4.9 | 46.7 ± 2.1 |
| 60 | 75.1 ± 4.1 | 72.9 ± 2.4 |
| 80 | 91.3 ± 1.2 | 82.3 ± 3.1 |
La sustitución de Cr por Al en la estructura M (acac) 3 podría ocurrir de manera aleatoria, es decir, un sitio metálico tiene la misma probabilidad de contener un átomo de aluminio o cromo. Alternativamente, si el cromo tuviera preferencia por sitios específicos, estaría presente una estructura súper reticular de menor simetría. Tal orden no se observa ya que todas las muestras no muestran reflexiones adicionales que las que pueden ser indexadas a la célula monoclínica. Por lo tanto, se puede concluir que el Al (acac) 3 y el Cr (acac) 3 de hecho forman soluciones sólidas: Al 1-x Cr x (acac) 3.
El análisis de microsonda electrónica, mediante espectrometría dispersiva de longitud de onda (WDS), en el cristal individual del que se recolectaron datos cristalográficos de rayos X proporciona la composición “real” de cada cristal. El análisis se realizó en al menos 6 sitios en cada cristal utilizando un punto de análisis de 10 μm que proporciona una medida de la homogeneidad dentro del cristal individual para el cual se recolectaron datos cristalográficos de rayos X. Un ejemplo de una imagen SEM de uno de los cristales y el análisis puntual se da en la Figura\(\PageIndex{2}\). Los datos de la Tabla\(\PageIndex{1}\) y la Figura\(\PageIndex{2}\) demuestran que si bien un lote de cristales puede contener cristales individuales con diferentes composiciones, cada cristal individual es en realidad razonablemente homogéneo. Hay, para la mayoría de las muestras, una varianza significativa entre la relación molar Al: Cr en el material a granel y un cristal individual elegido para difracción de rayos X. La variación en la relación Al:Cr dentro de cada cristal individual (± 10%) es mucho menor que la que existe entre los cristales.

Comparación de los métodos
Método 1
Dado que el Método 1 no refina el %Cr y se basa en un insumo para la composición porcentual de Al y Cr del material “a granel”, es decir, el %Cr en la masa total del material (Tabla\(\PageIndex{1}\), Columna 1), a diferencia del análisis del monocristal sobre el que se realizó la difracción de rayos X, (Tabla\(\PageIndex{1}\), Columna 2), cuanto más cercanos estaban estos valores al valor “real” determinado por WDS para el cristal sobre el que se realizó la difracción de rayos X (Tabla\(\PageIndex{1}\), Columna 1 vs 2) y luego más cerca está el refinamiento general de la estructura a los de los Métodos 2 - 4.
Si bien esta suposición es obviamente inválida para muchas de las muestras, es una que se usa a menudo cuando se dispone de datos masivos (por ejemplo, de RMN). Sin embargo, como no hay razón para suponer que un cristal es completamente representativo de la muestra a granel, no es prudente confiar únicamente en dichos datos.
Método 2
Este método siempre produjo valores finales, refinados, de ocupación que fueron cercanos a los obtenidos de WDS (Tabla\(\PageIndex{1}\)). Este enfoque supone que el movimiento de los átomos metálicos centrales es idéntico. Si bien esto obviamente no es estrictamente cierto ya que son de diferente tamaño, los resultados aquí obtenidos implican que se trata de una aproximación razonable donde se requieren datos de conectividad simples. Para muestras donde la cantidad de uno de los elementos (es decir, Cr) es muy baja por lo que a menudo no se puede obtener un buen refinamiento. En estos casos, al refinar los valores de ocupación, eso para Al superaría 1 mientras que el de Cr sería inferior a 1!
Método 3
En algunos casos, a pesar de la interrelación entre los parámetros de ocupación y desplazamiento, la convergencia se obtuvo con éxito. En estos casos las ocupaciones refinadas fueron ligeramente más cercanas a las observadas en EDS que los valores de ocupación obtenidos mediante el Método 2. Sin embargo, para algunas muestras con mayor contenido de Cr el refinamiento fue inestable y no convergería. No es seguro si esta observación se debió al aumento del porcentaje de Cr o simplemente a la menor calidad de los datos.
Si bien este método permite refinar cualquier diferencia en el movimiento atómico entre los dos metales, requiere datos de calidad extremadamente alta para que esta diferencia se determine de manera confiable.
Método 4
Este enfoque agrega poco a los resultados finales.
Correlación entre composición analizada y composición refinada
La Figura\(\PageIndex{3}\) muestra la relación entre la concentración de cromo (%Cr) determinada a partir de WDS y el refinamiento de los datos de difracción de rayos X usando los Métodos 2 o 3 (etiquetados en la Figura\(\PageIndex{3}\). Claramente existe una buena correlación, con solo una ligera divergencia a alta concentración de Cr. Esto es sin duda una consecuencia de tratar de refinar una fracción baja de un átomo de luz (Al) en presencia de una gran fracción de un átomo más pesado (Cr). La difracción de rayos X es, por lo tanto, un método preciso para determinar las relaciones M:M' en solución sólida cristalina.