
10.4: Espectroscopia de Absorción Atómica
- Page ID
- 75900

Guystav Kirchoff y Robert Bunsen utilizaron por primera vez la absorción atómica, junto con la emisión atómica, en 1859 y 1860 como medio para identificar átomos en llamas y gases calientes. Si bien la emisión atómica continuó desarrollándose como técnica analítica, el progreso en la absorción atómica languideció durante casi un siglo. La espectroscopia moderna de absorción atómica tiene sus inicios en 1955 como resultado del trabajo independiente de A. C. Walsh y C. T. J. Alkemade [(a) Walsh, A. Anal. Chem. 1991, 63, 933A—941A; b) Koirtyohann, S. R. Anal. Chem. 1991, 63, 1024A-1031A; c) Slavin, W. Anal. Chem. 1991, 63, 1033A-1038A]. Los instrumentos comerciales estaban en su lugar a principios de la década de 1960, y pronto se evidenció la importancia de la absorción atómica como técnica analítica.
Instrumentación
Los espectrofotómetros de absorción atómica utilizan la misma óptica de haz simple o doble haz descrita anteriormente para los espectrofotómetros de absorción molecular (ver Figura 10.3.2 y Figura 10.3.3). Sin embargo, existe una necesidad adicional importante en la espectroscopia de absorción atómica: primero debemos encubrir el analito en átomos libres. En la mayoría de los casos el analito está en forma de solución. Si la muestra es un sólido, entonces debemos llevar el analito a solución antes del análisis. Al analizar un sedimento lacustre para Cu, Zn y Fe, por ejemplo, llevamos los analitos en solución como Cu 2 +, Zn 2 + y Fe 3 + extrayéndolos con un reactivo adecuado. Por ello, en este capítulo sólo se considera la introducción de muestras de solución.
El reactivo que elegimos usar para poner un analito en solución depende de nuestros objetivos de investigación. Si necesitamos conocer la cantidad total de metal en el sedimento, entonces podríamos intentar una digestión por microondas usando una mezcla de ácidos concentrados, como HNO 3, HCl y HF. Esto destruye la matriz del sedimento y pone todo en solución. Por otro lado, si nuestro interés son los metales biológicamente disponibles, podríamos extraer la muestra en condiciones más suaves utilizando, por ejemplo, una solución diluida de HCl o CH 3 COOH a temperatura ambiente.
Atomización
El proceso de convertir un analito en un átomo gaseoso libre se llama atomización. La conversión de un analito acuoso en un átomo libre requiere que eliminemos el disolvente, volatilizemos el analito y, si es necesario, disociemos el analito en átomos libres. Desolvar una solución acuosa de CuCl 2, por ejemplo, nos deja con partículas sólidas de CuCl 2. La conversión del CuCl 2 particulado en átomos de fase gaseosa de Cu y Cl requiere energía térmica.
\[\mathrm{CuCl}_{2}(a q) \rightarrow \mathrm{CuCl}_{2}(s) \rightarrow \mathrm{Cu}(g)+2 \mathrm{Cl}(g) \nonumber\]
Existen dos métodos comunes de atomización: la atomización a la llama y la atomización electrotérmica, aunque algunos elementos se atomizan usando otros métodos.
Atomizador de llama
La figura 10.4.1 muestra un conjunto típico de atomización de llama con vistas en primer plano de varios componentes clave. En la unidad aquí mostrada, la muestra acuosa se introduce en el conjunto pasando una corriente de aire comprimido a alta presión más allá del extremo de un tubo capilar sumergido en la muestra. Cuando la muestra sale del nebulizador, choca con una perla de impacto de vidrio, lo que la convierte en una fina niebla de aerosol dentro de la cámara de pulverización. La niebla de aerosol es barrida a través de la cámara de pulverización por los gases de combustión, aire comprimido y acetileno en este caso, hasta la cabeza del quemador donde la energía térmica de la llama desolva la niebla del aerosol en un aerosol seco de partículas pequeñas y sólidas. La energía térmica de la llama volatiliza entonces las partículas, produciendo un vapor que consiste en especies moleculares, especies iónicas y átomos libres.

Quemador. El quemador de ranura en la Figura 10.4.1 a proporciona una longitud de ruta óptica larga y una llama estable. Debido a que la absorbancia es directamente proporcional a la longitud de la trayectoria, una longitud de trayectoria larga proporciona una mayor sensibilidad. Una llama estable minimiza la incertidumbre debida a las fluctuaciones en la llama.
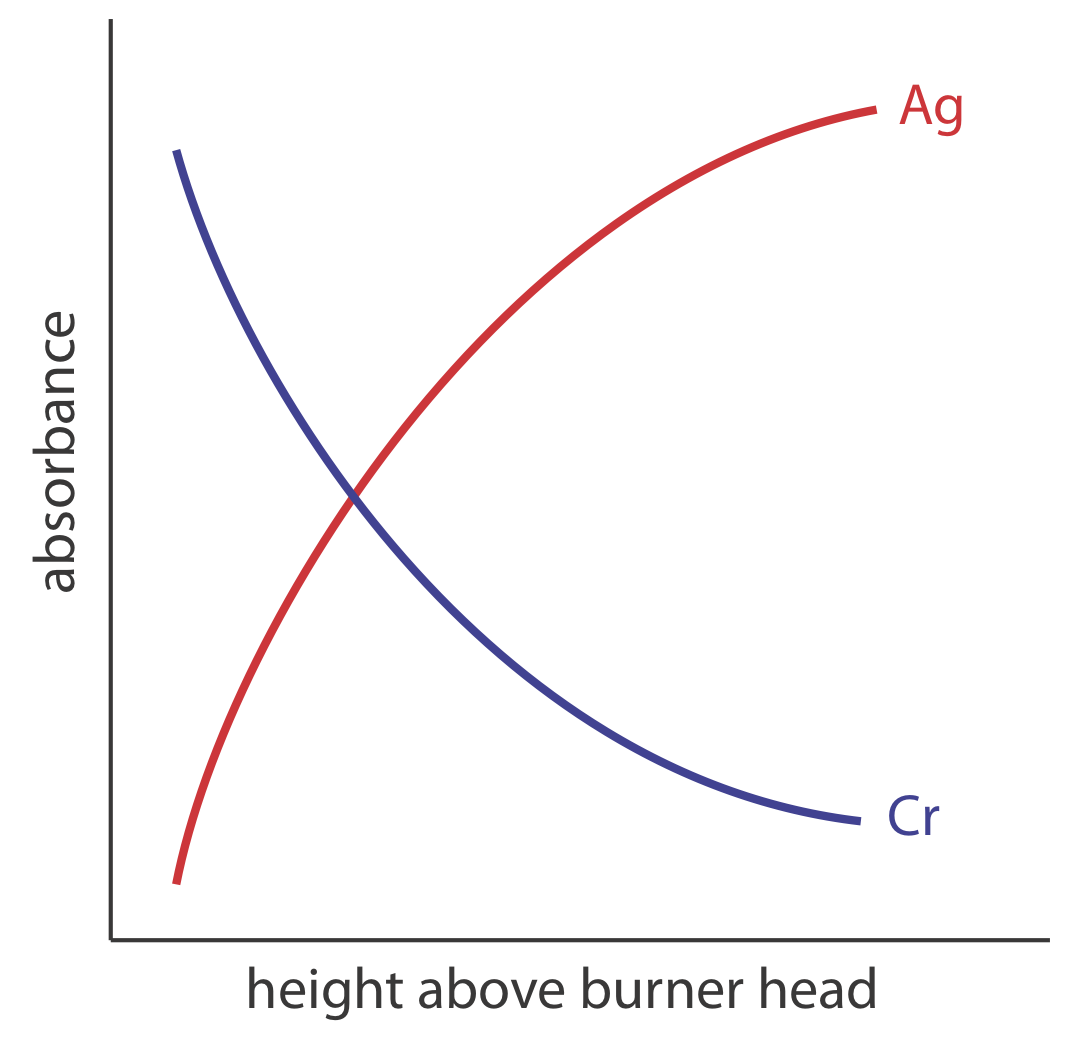
El quemador está montado en una plataforma ajustable que permite que todo el conjunto se mueva horizontal y verticalmente. Los ajustes horizontales aseguran que la llama esté alineada con la trayectoria óptica del instrumento. Los ajustes verticales cambian la altura dentro de la llama a partir de la cual se monitorea la absorbancia. Esto es importante porque dos procesos competidores afectan la concentración de átomos libres en la llama. Cuanto más tiempo pasa un analito en la llama, mayor es la eficiencia de atomización; así, la producción de átomos libres aumenta con la altura. Por otro lado, un mayor tiempo de residencia permite más oportunidades para que los átomos libres se combinen con el oxígeno para formar un óxido molecular. Como se ve en la Figura 10.4.2 , para un metal esto es fácil de oxidar, como el Cr, la concentración de átomos libres es mayor justo por encima de la cabeza del quemador. Para un metal, como el Ag, que es difícil de oxidar, la concentración de átomos libres aumenta de manera constante con la altura.
Llama. La temperatura de la llama, que afecta la eficiencia de la atomización, depende de la mezcla combustible-oxidante, varios ejemplos de los cuales se enumeran en la Tabla 10.4.1 . De estas, las llamas de aire, acetileno y óxido nitroso, acetileno son las más populares. Normalmente el combustible y el oxidante se mezclan en una relación aproximadamente estequiométrica; sin embargo, una mezcla rica en combustible puede ser necesaria para analitos fácilmente oxidados.
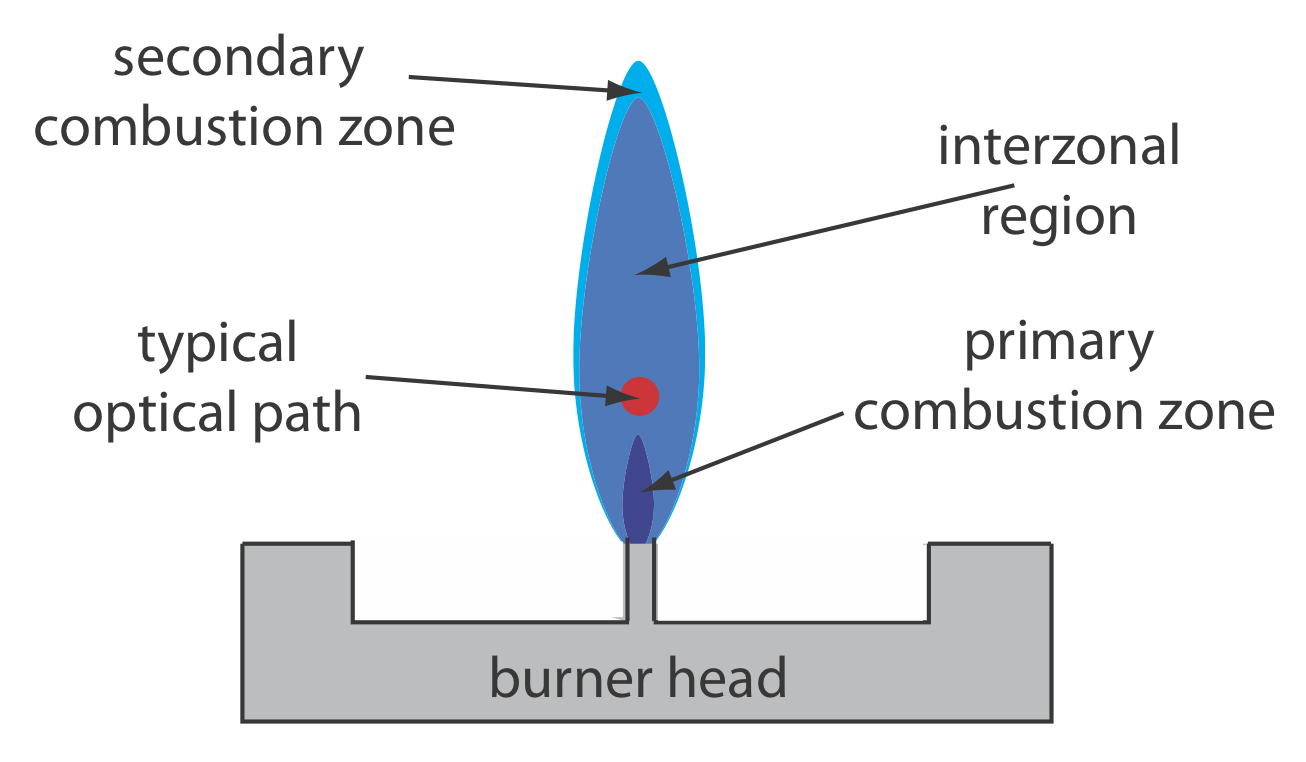
La Figura 10.4.3 muestra una sección transversal a través de la llama, mirando hacia abajo la trayectoria óptica de la radiación fuente. La zona de combustión primaria suele ser rica en productos de combustión de gas que emiten radiación, limitante es útil para la absorción atómica. La región interzonal generalmente es rica en átomos libres y proporciona la mejor ubicación para medir la absorción atómica. La parte más caliente de la llama suele estar entre 2 y 3 cm por encima de la zona de combustión primaria. A medida que los átomos se acercan a la zona de combustión secundaria de la llama, la disminución de la temperatura permite la formación de especies moleculares estables.
Introducción a la muestra. El medio más común para introducir una muestra en un atomizador de llama es una aspiración continua en la que la muestra fluye a través del quemador mientras monitoreamos la absorbancia. La aspiración continua es intensiva de muestra, requiriendo típicamente de 2—5 mL de muestra.
El micromuestreo de llama nos permite introducir una muestra discreta de volumen fijo, y es útil si tenemos una cantidad limitada de muestra o cuando la matriz de la muestra es incompatible con el atomizador de llama. Por ejemplo, la aspiración continua de una muestra que tiene una alta concentración de sólidos disueltos —agua de mar, por ejemplo, viene a la mente— puede generar una desposición sólida en la cabeza del quemador que obstruye la llama y que disminuye la absorbancia. El micromuestreo de llama se realiza usando una micropipeta para colocar 50-250 μL de muestra en un embudo de teflón conectado al nebulizador, o sumergiendo el tubo del nebulizador en la muestra por un corto tiempo. El muestreo por inmersión generalmente se realiza con un muestreador automático. La señal para el micromuestreo de llama es un pico transitorio cuya altura o área es proporcional a la cantidad de analito que se inyecta.
Ventajas y Desventajas de la Atomización de Llama. La principal ventaja de la atomización a la llama es la reproducibilidad con la que se introduce la muestra en el espectrofotómetro; una desventaja significativa es que la eficiencia de la atomización es bastante pobre. Hay dos razones para una baja eficiencia de atomización. Primero, la mayoría de las gotitas de aerosol producidas durante la nebulización son demasiado grandes para ser transportadas a la llama por los gases de combustión. En consecuencia, hasta el 95% de la muestra nunca llega a la llama, que es la razón de la línea de desechos que se muestra en la parte inferior de la cámara de pulverización en la Figura 10.4.1 . Una segunda razón para la baja eficiencia de atomización es que el gran volumen de gases de combustión diluye significativamente la muestra. En conjunto, estas contribuciones a la eficiencia de la atomización reducen la sensibilidad debido a que la concentración del analito en la llama puede ser un factor\(2.5 \times 10^{-6}\) menor que la de la solución [Ingle, J. D.; Crouch, S. R. Spectrochemical Analysis, Prentice-Hall: Englewood Cliffs, NJ, 1988; p. 275].
Atomizadores Electrotérmicos
Se logra una mejora significativa en la sensibilidad mediante el uso del calentamiento resistivo de un tubo de grafito en lugar de una llama. Un atomizador electrotérmico típico, también conocido como horno de grafito, consiste en un tubo de grafito cilíndrico de aproximadamente 1—3 cm de longitud y 3—8 mm de diámetro. Como se muestra en la Figura 10.4.4 , el tubo de grafito se aloja en un conjunto sellado que tiene una ventana ópticamente transparente en cada extremo. A través del horno se pasa una corriente continua de gas inerte, que protege el tubo de grafito de la oxidación y elimina los productos gaseosos producidos durante la atomización. Se utiliza una fuente de alimentación para pasar una corriente a través del tubo de grafito, lo que resulta en calentamiento resistivo.
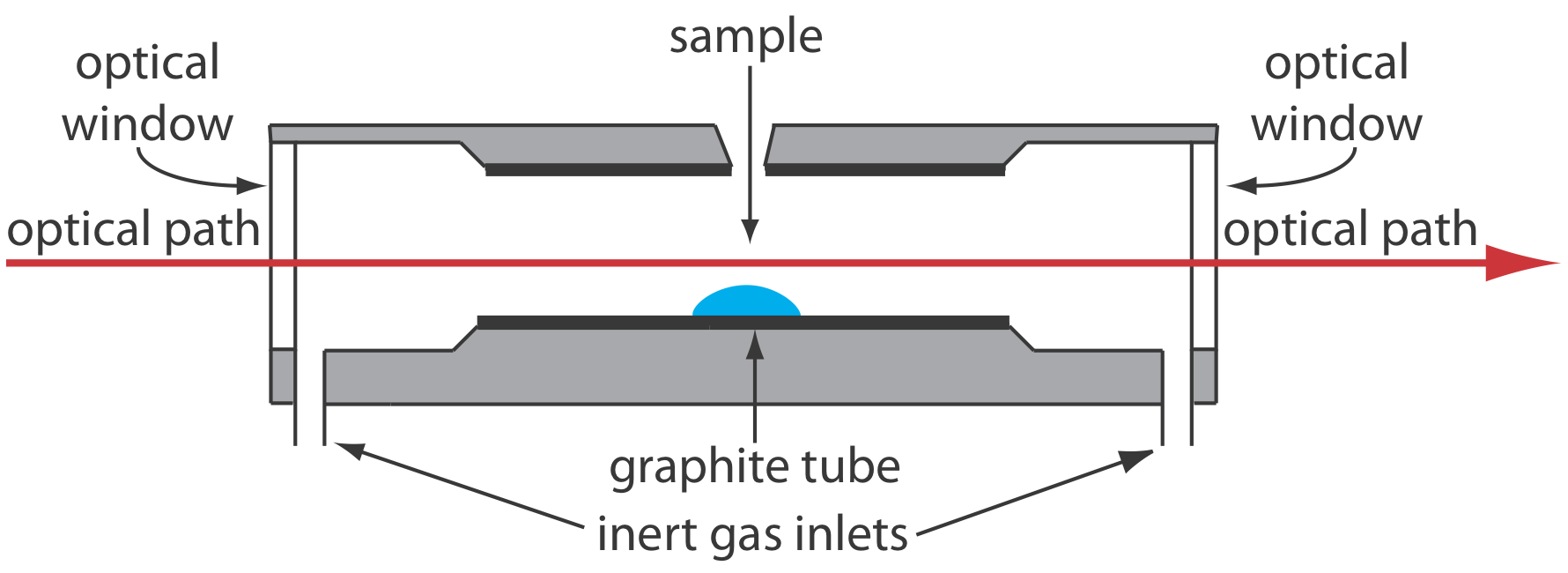
Muestras de entre 5—50 μL se inyectan en el tubo de grafito a través de un pequeño orificio en la parte superior del tubo. La atomización se logra en tres etapas. En la primera etapa la muestra se seca a un residuo sólido utilizando una corriente que eleva la temperatura del tubo de grafito a aproximadamente 110 o C. En la segunda etapa, que se denomina ceniza, la temperatura se incrementa a entre 350—1200 o C. A estas temperaturas el material orgánico en el se convierte en CO 2 y H 2 O, y los materiales inorgánicos volátiles se vaporizan. Estos gases son eliminados por el flujo de gas inerte. En la etapa final la muestra se atomiza aumentando rápidamente la temperatura hasta entre 2000—3000 o C. El resultado es un pico de absorbancia transitoria cuya altura o área es proporcional a la cantidad absoluta de analito inyectado en el tubo de grafito. En conjunto, las tres etapas toman aproximadamente 45—90 s, siendo la mayor parte de este tiempo utilizadas para secar y triturar la muestra.
La atomización electrotérmica proporciona una mejora significativa en la sensibilidad al retener el analito gaseoso en el pequeño volumen dentro del tubo de grafito. La concentración del analito en la fase vapor resultante es tanto\(1000 \times\) mayor que en una atomización a la llama [Parsons, M. L.; Major, S.; Forster, A. R. Appl. Espectrosc. 1983, 37, 411—418]. Esta mejora en la sensibilidad, y la consiguiente mejora en los límites de detección, se compensa con una disminución significativa de la precisión. La eficiencia de atomización está fuertemente influenciada por el contacto de la muestra con el tubo de grafito, el cual es difícil de controlar reproduciblemente.
Varios métodos de atomización
Algunos elementos se atomizan mediante el uso de una reacción química para producir un producto volátil. Elementos como As, Se, Sb, Bi, Ge, Sn, Te y Pb, por ejemplo, forman hidruros volátiles cuando reaccionan con NaBH 4 en presencia de ácido. Un gas inerte transporta el hidruro volátil a una llama o a un tubo de observación de cuarzo calentado situado en la trayectoria óptica. El mercurio se determina por el método de vapor frío en el que se reduce a mercurio elemental con SnCl 2. El Hg volátil es transportado por un gas inerte a un tubo de observación sin calentar situado en la trayectoria óptica del instrumento.
Aplicaciones Cuantitativas
La absorción atómica se usa ampliamente para el análisis de metales traza en una variedad de matrices de muestra. Usando Zn como ejemplo, existen métodos estándar de absorción atómica para su determinación en muestras tan diversas como agua y aguas residuales, aire, sangre, orina, tejido muscular, cabello, leche, cereales para el desayuno, champús, aleaciones, baños industriales de chapado, gasolina, aceite, sedimentos y rocas.
El desarrollo de un método cuantitativo de absorción atómica requiere varias consideraciones, entre ellas elegir un método de atomización, seleccionar la longitud de onda y el ancho de la hendidura, preparar la muestra para su análisis, minimizar las interferencias espectrales y químicas y seleccionar un método de estandarización. Cada uno de estos temas es considerado en esta sección.
Desarrollo de un Método Cuantitativo
¿Atomización Electrotérmica o Llama? El factor más importante para elegir un método de atomización es la concentración del analito. Debido a su mayor sensibilidad, se necesita menos analito para lograr una absorbancia dada cuando se utiliza la atomización electrotérmica. La Tabla 10.4.2 , que compara la cantidad de analito necesaria para lograr una absorbancia de 0.20 al usar atomización de llama y atomización electrotérmica, es útil a la hora de seleccionar un método de atomización. Por ejemplo, la atomización por llama es el método de elección si nuestras muestras contienen 1—10 mg de Zn 2 + /L, pero la atomización electrotérmica es la mejor opción para muestras que contienen 1—10 μg de Zn 2 + /L.
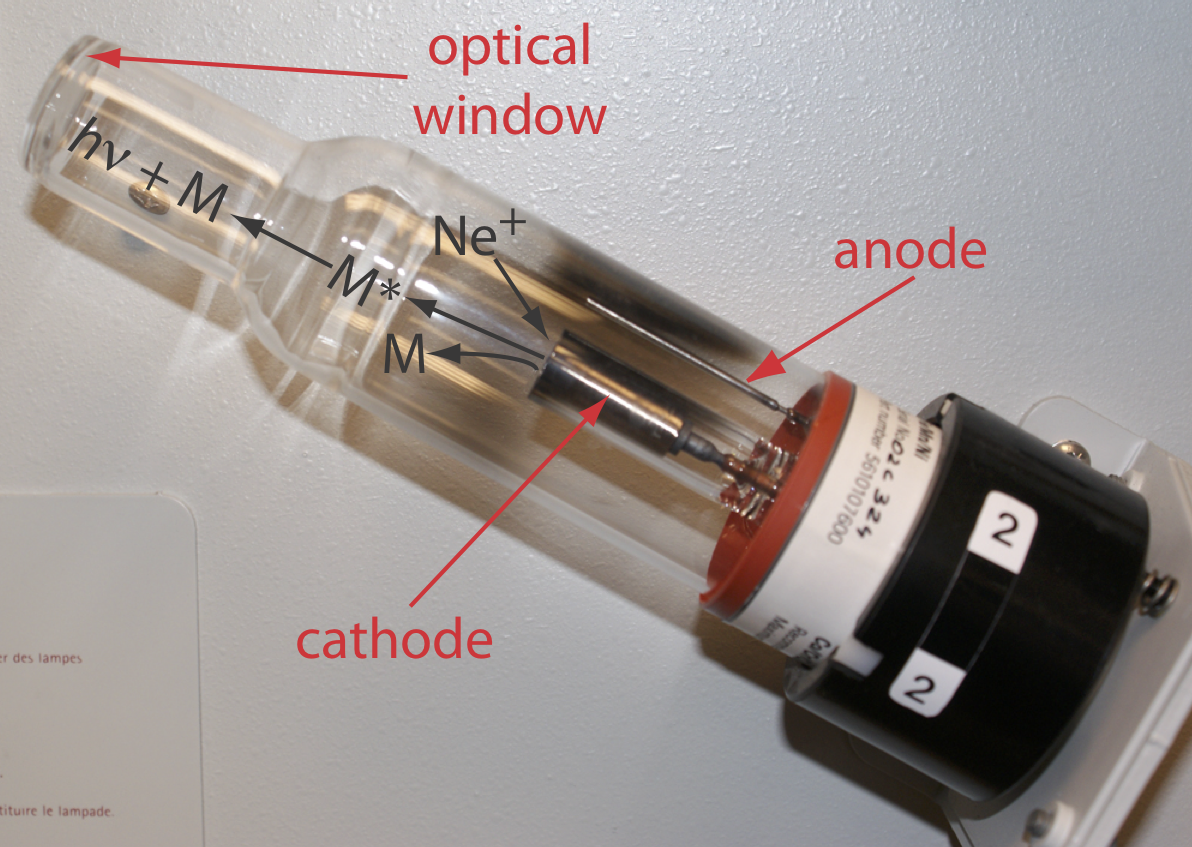
Seleccionar la longitud de onda y el ancho de hendidura. La fuente de absorción atómica es una lámpara de cátodo hueco que consiste en un cátodo y ánodo encerrados dentro de un tubo de vidrio lleno de una baja presión de un gas inerte, como Ne o Ar (Figura 10.4.5 ). La aplicación de un potencial a través de los electrodos ioniza el gas de relleno. Los iones de gas cargados positivamente chocan con el cátodo cargado negativamente, pulverizando átomos de la superficie del cátodo. Algunos de los átomos pulverizados se encuentran en estado excitado y emiten radiación característica del metal o metales a partir de los cuales se fabrica el cátodo. Al modelar el cátodo a partir del analito metálico, una lámpara de cátodo hueco proporciona líneas de emisión que corresponden al espectro de absorción del analito.
Debido a que las líneas de absorción atómica son estrechas, necesitamos usar una fuente de línea en lugar de una fuente continua (compare, por ejemplo, la Figura 10.2.4 con la Figura 10.2.6). El ancho de banda efectivo cuando se usa una fuente continua es aproximadamente\(1000 \times\) mayor que una línea de absorción atómica; así, P T ≈ P 0,% T ≈ 100 y A ≈ 0. Debido a que una lámpara de cátodo hueco es una fuente de línea, P T y P 0 tienen valores diferentes dando un% T < 100 y A > 0.
Cada elemento en una lámpara de cátodo hueco proporciona varias líneas de emisión atómica que podemos utilizar para la absorción atómica. Por lo general, la longitud de onda que proporciona la mejor sensibilidad es la que elegimos usar, aunque una longitud de onda menos sensible puede ser más apropiada para una muestra que tenga mayor concentración de analito. Para la lámpara de cátodo hueco de Cr en la Tabla 10.4.3 , la mejor sensibilidad se obtiene usando una longitud de onda de 357.9 nm.
Otra consideración es la intensidad de la línea de emisión. Si varias líneas de emisión cumplen con nuestros requisitos de sensibilidad, es posible que deseemos usar la línea de emisión con el P 0 relativo más grande porque hay menos incertidumbre en la medición de P 0 y P T. Al analizar una muestra que es ≈10 mg Cr/L, por ejemplo, las tres primeras longitudes de onda en la Tabla 10.4.3 proporcionan una sensibilidad apropiada; las longitudes de onda de 425.4 nm y 429.0 nm, sin embargo, tienen un P 0 mayor y proporcionarán menos incertidumbre en la absorbancia medida.
El espectro de emisión para una lámpara de cátodo hueco incluye, además de las líneas de emisión del analito, líneas de emisión adicionales de impurezas presentes en el cátodo metálico y del gas de relleno. Estas líneas adicionales son una fuente potencial de radiación parásita que podría resultar en una desviación instrumental de la ley de Beer. El ancho de hendidura del monocromador se establece lo más ancho posible para mejorar el rendimiento de la radiación y lo suficientemente estrecho como para eliminar estas fuentes de radiación parásita.
Preparación de la Muestra. La llama y la atomización electrotérmica requieren que el analito esté en solución. Las muestras sólidas se ponen en solución disolviéndolas en un disolvente apropiado. Si la muestra no es soluble se digiere, ya sea en placa caliente o por microondas, usando HNO 3, H 2 SO 4, o HClO 4. Alternativamente, podemos extraer el analito usando un extractor Soxhlet. Las muestras líquidas se analizan directamente o se extraen los analitos si la matriz es compatible con el método de atomización. Una muestra de suero, por ejemplo, es difícil de aspirar cuando se usa atomización por llama y puede producir una absorbancia de fondo inaceptablemente alta cuando se usa atomización electrotérmica. Una extracción líquido-líquido usando un solvente orgánico y un agente quelante se usa frecuentemente para concentrar analitos. Las soluciones diluidas de Cd 2 +, Co 2+, Cu 2 +, Fe 3+, Pb 2 +, Ni 2 + y Zn 2 +, por ejemplo, se concentran extrayendo con una solución de amonio ditiocarbamato de pirrolidina en metil isobutil cetona.
Minimizar la interferencia espectral. Una interferencia espectral ocurre cuando la línea de absorción de un analito se solapa con la línea o banda de absorción de un interferente. Debido a que son tan estrechos, la superposición de dos líneas de absorción atómica rara vez es un problema. Por otro lado, la amplia banda de absorción de una molécula o la dispersión de la radiación fuente es una interferencia espectral potencialmente grave.
Una consideración importante cuando se usa una llama como fuente de atomización es su efecto sobre la absorbancia medida. Entre los productos de combustión se encuentran especies moleculares que exhiben amplias bandas de absorción y partículas que dispersan la radiación de la fuente. Si no conseguimos compensar estas interferencias espectrales, entonces la intensidad de la radiación transmitida es menor de lo esperado. El resultado es un aumento aparente en la absorbancia de la muestra. Afortunadamente, la absorción y dispersión de la radiación por la llama se corrigen analizando un blanco.
Las interferencias espectrales también ocurren cuando componentes de la matriz de la muestra distintos del analito reaccionan para formar especies moleculares, como óxidos e hidróxidos. La absorción y dispersión resultante constituye el fondo de la muestra y puede presentar un problema significativo, particularmente en longitudes de onda inferiores a 300 nm donde la dispersión de la radiación se vuelve más importante. Si conocemos la composición de la matriz de la muestra, entonces podemos preparar nuestras muestras usando una matriz idéntica. En este caso la absorción de fondo es la misma tanto para las muestras como para los estándares. Alternativamente, si el fondo se debe a un componente de matriz conocido, entonces podemos agregar ese componente en exceso a todas las muestras y estándares para que la contribución del interferente natural sea insignificante. Finalmente, muchas interferencias debidas a la matriz de la muestra se eliminan al aumentar la temperatura de atomización. Por ejemplo, cambiar a una llama de mayor temperatura ayuda a evitar la formación de óxidos e hidróxidos interferentes.
Si se desconoce la identidad de la interferencia de la matriz, o si no es posible ajustar las condiciones de la llama o del horno para eliminar la interferencia, entonces debemos encontrar otro método para compensar la interferencia de fondo. Se han desarrollado varios métodos para compensar las interferencias de la matriz, y la mayoría de los espectrofotómetros de absorción atómica incluyen uno o más de estos métodos.
Uno de los métodos más comunes para la corrección de fondo es utilizar una fuente continua, como una lámpara D 2. Debido a que una lámpara D 2 es una fuente continua, la absorbancia de su radiación por la línea de absorción estrecha del analito es insignificante. Sólo el fondo, por lo tanto, absorbe la radiación de la lámpara D 2. Tanto el analito como el fondo, por otro lado, absorben la radiación del cátodo hueco. Al restar la absorbancia para la lámpara D 2 de la de la lámpara de cátodo hueco se obtiene una absorbancia corregida que compensa la interferencia de fondo. Aunque este método de corrección de fondo es efectivo, se supone que la absorbancia de fondo es constante en el rango de longitudes de onda pasadas por el monocromador. Si esto no es cierto, entonces restando las dos absorbancias subestima o sobreestima el fondo.
Se han desarrollado otros métodos de corrección de fondo, incluyendo la corrección de fondo del efecto Zeeman y la corrección de fondo Smith—Hieftje, ambos incluidos en algunos espectrofotómetros de absorción atómica disponibles comercialmente. Consulte los recursos adicionales del capítulo para obtener información adicional.
Minimizar las interferencias químicas. El análisis cuantitativo de algunos elementos se complica por las interferencias químicas que ocurren durante la atomización. Las interferencias químicas más comunes son la formación de compuestos no volátiles que contienen el analito y la ionización del analito.
Un ejemplo de la formación de un compuesto no volátil es el efecto de\(\text{PO}_4^{3-}\) o Al 3 + en el análisis de absorción atómica de llama de Ca 2 +. En un estudio, por ejemplo, agregar 100 ppm Al 3 + a una solución de 5 ppm Ca 2 + disminuyó la absorbancia de iones calcio de 0.50 a 0.14, mientras que agregar 500 ppm\(\text{PO}_4^{3-}\) a una solución similar de Ca 2 + disminuyó la absorbancia de 0.50 a 0.38. Estas interferencias se atribuyen a la formación de partículas no volátiles de Ca 3 (PO 4) 2 y un óxido de Al—Ca—O [Hosking, J. W.; Snell, N. B.; Sturman, B. T. J. Chem. Educ. 1977, 54, 128—130].
Al usar la atomización de llama, podemos minimizar la formación de compuestos no volátiles aumentando la temperatura de la llama cambiando la relación combustible a oxidante o cambiando a una combinación diferente de combustible y oxidante. Otro enfoque es agregar un agente liberador o un agente protector a la muestra. Un agente de arrendamiento es una especie que reacciona preferentemente con el interferente, liberando el analito durante la atomización. Por ejemplo, Sr 2 + y La 3 + sirven como agentes liberadores para el análisis de Ca 2 + en presencia de\(\text{PO}_4^{3-}\) o Al 3 +. Al agregar 2000 ppm de SrCl 2 a las mezclas de Ca 2 +/\(\text{PO}_4^{3-}\)y Ca 2 + /Al 3+ descritas en el párrafo anterior se incrementó la absorbancia a 0.48. Un agente protector reacciona con el analito para formar un complejo volátil estable. Al agregar 1% w/w EDTA a la\(\text{PO}_4^{3-}\) solución de Ca 2 +/descrita en el párrafo anterior se incrementó la absorbancia a 0.52.
Una interferencia de ionización ocurre cuando la energía térmica de la llama o el atomizador electrotérmico es suficiente para ionizar el analito
\[\mathrm{M}(s)\rightleftharpoons \ \mathrm{M}^{+}(a q)+e^{-} \label{10.1}\]
donde M es el analito. Debido a que los espectros de absorción para M y M + son diferentes, la posición del equilibrio en la reacción\ ref {10.1} afecta la absorbancia a longitudes de onda donde M absorbe. Para limitar la ionización agregamos una alta concentración de un supresor de ionización, que es una especie que ioniza más fácilmente que el analito. Si la concentración del supresor de ionización es suficiente, entonces la mayor concentración de electrones en la llama empuja la reacción\ ref {10.1} hacia la izquierda, impidiendo la ionización del analito. El potasio y el cesio se utilizan frecuentemente como supresores de ionización debido a su baja energía de ionización.
Estandarización del Método. Como la ley de Beer también se aplica a la absorción atómica, podríamos esperar que las curvas de calibración de absorción atómica sean lineales. En la práctica, sin embargo, la mayoría de las curvas de calibración de absorción atómica son no lineales o lineales en un rango limitado de concentraciones. La no linealidad en la absorción atómica es consecuencia de limitaciones instrumentales, incluyendo la radiación parásita de la lámpara de cátodo hueco y la variación en la absortividad molar a través de la línea de absorción. El trabajo cuantitativo preciso, por lo tanto, requiere un medio adecuado para calcular la curva de calibración a partir de un conjunto de estándares.
Cuando es posible, se realiza mejor un análisis cuantitativo utilizando estándares externos. Desafortunadamente, las interferencias matriciales son un problema frecuente, particularmente cuando se utiliza la atomización electrotérmica. Por esta razón, a menudo se utiliza el método de adiciones estándar. Una limitación a este método de estandarización, sin embargo, es el requisito de una relación lineal entre absorbancia y concentración.
La mayoría de los instrumentos incluyen varios algoritmos diferentes para calcular la curva de calibración. El instrumento en mi laboratorio, por ejemplo, incluye cinco algoritmos. Tres de los algoritmos ajustan los datos de absorbancia usando funciones polinómicas lineales, cuadráticas o cúbicas de la concentración del analito. También incluye dos algoritmos que ajustan las concentraciones de los estándares a las funciones cuadráticas de la absorbancia.
Método Representativo 10.4.1: Determinación de Cu y Zn en Muestras de Tejido
La mejor manera de apreciar los detalles teóricos y prácticos discutidos en esta sección es examinar cuidadosamente un método analítico típico. Aunque cada método es único, la siguiente descripción de la determinación de Cu y Zn en tejidos biológicos proporciona un ejemplo instructivo de un procedimiento típico. La descripción aquí se basa en Bhattacharya, S. K.; Goodwin, T. G.; Crawford, A. J. Anal. Lt. 1984, 17, 1567—1593, y Crawford, A. J.; Bhattacharya, S. K. Instrumentos Varian en el Trabajo, número AA—46, abril de 1985.
Descripción del Método.
El cobre y el zinc se aíslan de las muestras de tejido digiriendo la muestra con HNO 3 después de eliminar primero cualquier tejido graso. La concentración de cobre y zinc en el sobrenadante se determina por absorción atómica usando una llama aire-acetileno.
Procedimiento.
Las muestras de tejido se obtienen mediante una biopsia con aguja muscular y se secan durante 24—30 h a 105 o C para eliminar todas las trazas de humedad. El tejido graso en una muestra seca se retira extrayendo durante la noche con éter anhidro. Después de retirar el éter, la muestra se seca para obtener el peso de tejido seco libre de grasa (FFDT). La muestra se digiere a 68 o C por 20—24 h usando 3 mL de HNO 3 0.75 M. Después de centrifugar a 2500 rpm por 10 minutos, el sobrenadante se transfiere a un matraz aforado de 5 ml. La digestión se repite dos veces más, durante 2—4 horas cada una, utilizando alícuotas de 0.9-mL de HNO 3 0.75 M. Estos sobrenadantes se agregan al matraz aforado de 5 mL, el cual se diluye a volumen con HNO 3 0.75 M. Las concentraciones de Cu y Zn en el sobrenadante diluido se determinan mediante espectroscopía de absorción atómica de llama usando una llama de aire-acetileno y estándares externos. El cobre se analiza a una longitud de onda de 324.8 nm con un ancho de hendidura de 0.5 nm, y el zinc se analiza a 213.9 nm con un ancho de ranura de 1.0 nm. La corrección de fondo usando una lámpara D 2 es necesaria para el zinc. Los resultados se reportan como μg de Cu o Zn por gramo de FFDT.
Preguntas.
1. Describir la matriz apropiada para los estándares externos y para el blanco?
La matriz para los estándares y el blanco deben coincidir con la matriz de las muestras; así, una matriz apropiada es 0.75 M HNO 3. Cualquier interferencia de otros componentes de la matriz de muestra se minimiza mediante la corrección de fondo.
2. ¿Por qué es necesaria una corrección de fondo para el análisis de Zn, pero no para el análisis de Cu?
La corrección de fondo compensa la absorción y dispersión de fondo debido a los interferentes en la muestra. Dichas interferencias son más severas cuando se usa una longitud de onda menor a 300 nm. Este es el caso del Zn, pero no para el Cu.
3. Una lámpara de cátodo hueco de Cu tiene varias líneas de emisión, cuyas propiedades se muestran en la siguiente tabla. Explica por qué este método utiliza la línea a 324.8 nm.
| longitud de onda (nm) | ancho de hendidura (nm) | mg Cu/L para A = 0.20 | P 0 (relativo) |
|---|---|---|---|
| 217.9 | 0.2 | 15 | 3 |
| 218.2 | 0.2 | 15 | 3 |
| 222.6 | 0.2 | 60 | 5 |
| 244.2 | 0.2 | 400 | 15 |
| 249.2 | 0.5 | 200 | 24 |
| 324.8 | 0.5 | 1.5 | 100 |
| 327.4 | 0.5 | 3 | 87 |
Con 1.5 mg Cu/L dando una absorbancia de 0.20, la línea de emisión a 324.8 nm tiene la mejor sensibilidad. Además, es la línea de emisión más intensa, lo que disminuye la incertidumbre en la absorbancia medida.
Para evaluar el método descrito en el Método Representativo 10.4.1, se prepara y analiza una serie de estándares externos, proporcionando los resultados que aquí se muestran [Crawford, A. J.; Bhattacharya, S. K. “Microanalysis of Copper and Zinc in Biopsy-Sized Tissue Spectrroscopy by Atomic Absorption Spectroscopy Using a Llama estequiométrica de aire-acetileno”, Varian Instruments at Work, número AA—46, abril de 1985].
| µg Cu/ml | absorbancia | µg Cu/ml | absorbancia |
|---|---|---|---|
| 0.000 | 0.000 | 0.500 | 0.033 |
| 0.100 | 0.006 | 0.600 | 0.039 |
| 0.200 | 0.013 | 0.700 | 0.046 |
| 0.300 | 0.020 | 1.00 | 0.066 |
| 0.400 | 0.026 |
Se utiliza un material de referencia estándar de hígado bovino para evaluar la precisión del método. Después de secar y extraer la muestra, una muestra de tejido FFDT de 11.23 mg da una absorbancia de 0.023. Reportar la cantidad de cobre en la muestra como μg Cu/g FFDT.
Solución
La regresión lineal de la absorbancia frente a la concentración de Cu en los estándares da la curva de calibración que se muestra a continuación y la siguiente ecuación de calibración.
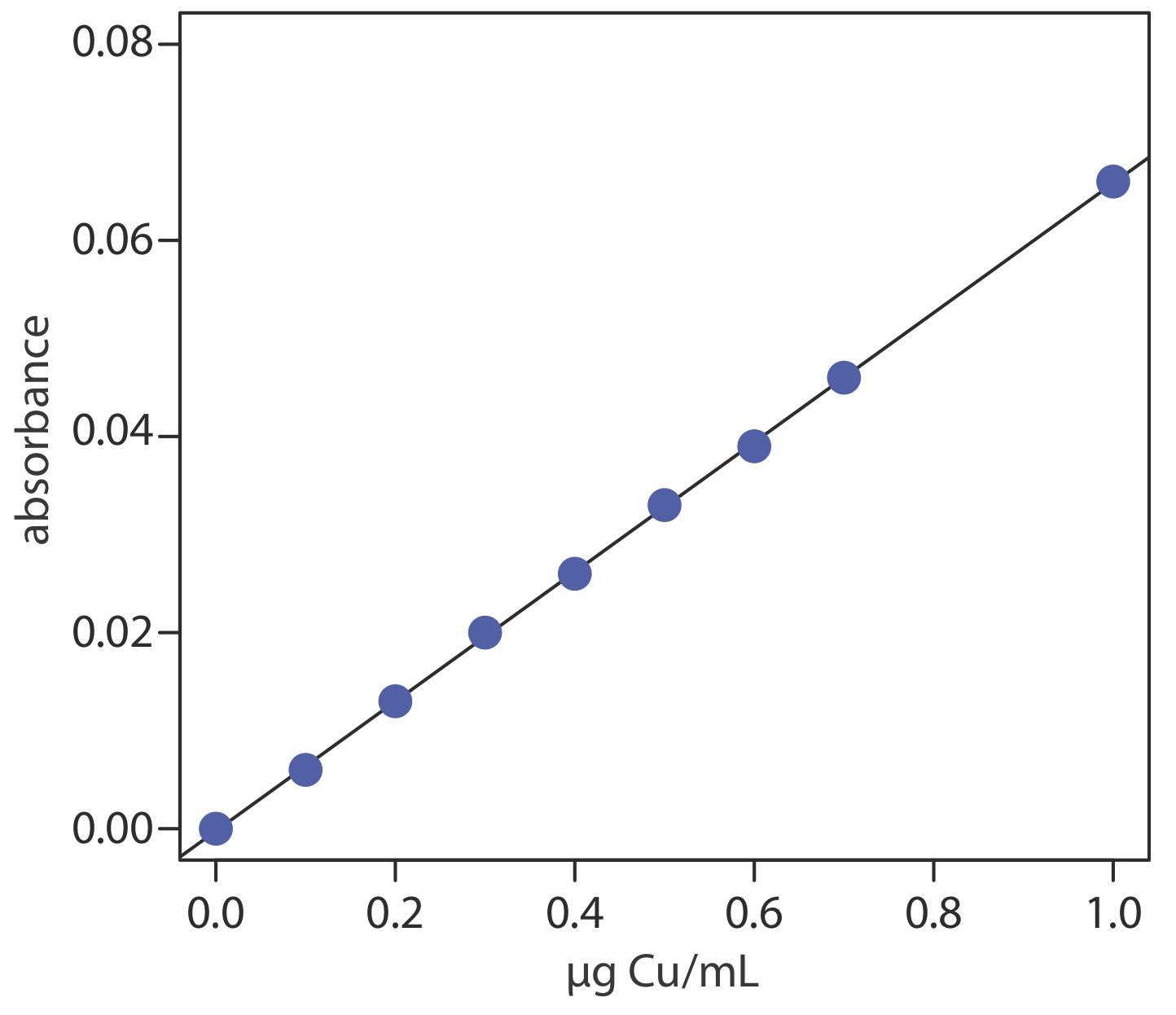
\[A=-0.0002+0.0661 \times \frac{\mu \mathrm{g} \ \mathrm{Cu}}{\mathrm{mL}} \nonumber\]
Sustituyendo la absorbancia de la muestra en la ecuación de calibración da la concentración de cobre como 0.351 μg/mL. La concentración de cobre en la muestra de tejido, por lo tanto, es
\[\frac { \frac{0.351 \mu \mathrm{g} \ \mathrm{Cu}}{\mathrm{mL}} \times 5.000 \ \mathrm{mL}} {0.01123 \text{ g sample}}=156 \ \mu \mathrm{g} \ \mathrm{Cu} / \mathrm{g} \ \mathrm{FDT} \nonumber\]
Evaluación de Espectroscopia de Absorción Atómica
Escala de Operación
La espectroscopia de absorción atómica es ideal para el análisis de analitos de traza y ultratraza, particularmente cuando se utiliza la atomización electrotérmica. Para analitos menores y mayores, la muestra se diluye antes del análisis. La mayoría de los análisis utilizan una macro o una muestra meso. El pequeño volumen requerido para la atomización electrotérmica o para el micromuestreo de llama, sin embargo, hace práctico el análisis de muestras micro y ultramicro.
Precisión
Si se minimizan las interferencias espectrales y químicas, se puede lograr rutinariamente una precisión de 0.5— 5%. Cuando la curva de calibración es no lineal, se mejora la precisión mediante el uso de un par de patrones cuyas absorbancias se ajustan estrechamente a la absorbancia de la muestra y asumiendo que el cambio en la absorbancia es lineal en este rango de concentración limitado. Los errores determinados para la atomización electrotérmica a menudo son mayores que los obtenidos con la atomización a la llama debido a interferencias de matriz más graves.
Precisión
Para una absorbancia mayor a 0.1—0.2, la desviación estándar relativa para la absorción atómica es 0.3— 1% para la atomización de llama y 1— 5% para la atomización electrotérmica. La principal limitación es la incertidumbre en la concentración de átomos de analito libres que resultan de las variaciones en la velocidad de aspiración, nebulización y atomización para un atomizador de llama, y la consistencia de las muestras inyectadas para la atomización electrotérmica.
Sensibilidad
La sensibilidad de un análisis de absorción atómica de llama está influenciada por la composición de la llama y por la posición en la llama desde la cual monitoreamos la absorbancia. Normalmente la sensibilidad de un análisis se optimiza aspirando una solución estándar de analito y ajustando la relación combustible a oxidante, el caudal del nebulizador y la altura del quemador, para dar la mayor absorbancia. Con la atomización electrotérmica, la sensibilidad se ve influenciada por las etapas de secado y cenizas que preceden a la atomización. La temperatura y el tiempo en cada etapa se optimizan para cada tipo de muestra.
La sensibilidad también está influenciada por la matriz de la muestra. Ya señalamos, por ejemplo, que la sensibilidad se ve disminuida por una interferencia química. Se puede lograr un aumento en la sensibilidad añadiendo un alcohol, éster o cetona de bajo peso molecular a la solución, o usando un disolvente orgánico.
Selectividad
Debido al estrecho ancho de las líneas de absorción, la absorción atómica proporciona una excelente selectividad. La absorción atómica se utiliza para el análisis de más de 60 elementos a concentraciones en o por debajo del nivel de μg/L.
Tiempo, Costo y Equipo
El tiempo de análisis cuando se utiliza la atomización por llama es corto, con rendimientos de muestra de 250—350 determinaciones por hora cuando se utiliza un sistema completamente automatizado. La atomización electrotérmica requiere sustancialmente más tiempo por análisis, con rendimientos máximos de muestra de 20 a 30 determinaciones por hora. El costo de un nuevo instrumento oscila entre $10,000 y $50,000 para la atomización por llama y desde $18,000—$70,000 para la atomización electrotérmica. Los instrumentos más caros en cada rango de precios incluyen óptica de doble haz, muestreadores automáticos y se pueden programar para análisis multielemental al permitir que la longitud de onda y la lámpara de cátodo hueco se cambien automáticamente.