
10.3: Espectroscopia UV/Vis e IR
- Page ID
- 75910

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)En la Figura 10.1.9 examinamos el método original de Nessler para hacer coincidir el color de una muestra con el color de un patrón. La coincidencia de colores es un proceso intensivo para el analista y, como era de esperar, los métodos espectroscópicos de análisis tardaron en encontrar el favor. En las décadas de 1930 y 1940 se introdujeron transductores fotoeléctricos para radiación ultravioleta y visible, y termopares para radiación infrarroja. Como resultado, la instrumentación moderna para espectroscopía de absorción estuvo disponible de manera rutinaria en la década de 1940; desde entonces, el progreso adicional ha sido rápido.
Instrumentación
Frecuentemente, un analista debe seleccionar entre varios instrumentos de diseño diferente, el instrumento más adecuado para un análisis particular. En esta sección se examinan diversos instrumentos para la espectroscopia de absorción molecular, con énfasis en sus ventajas y limitaciones. Los métodos de introducción de la muestra también se tratan en esta sección.
Diseños de Instrumentos para Absorción Molecular UV/Vis
Fotómetro de Filtro. El instrumento más simple para la absorción molecular de UV/Vis es un fotómetro de filtro (Figura 10.3.1 ), que utiliza un filtro de absorción o interferencia para aislar una banda de radiación. El filtro se coloca entre la fuente y la muestra para evitar que la muestra se descomponga cuando se expone a radiación de mayor energía. Un fotómetro de filtro tiene una única trayectoria óptica entre la fuente y el detector, y se llama instrumento de haz único. El instrumento se calibra a 0% T mientras se usa un obturador para bloquear la radiación de la fuente del detector. Después de abrir el obturador, el instrumento se calibra al 100% T usando una pieza en blanco apropiada. Luego se reemplaza el blanco con la muestra y se mide su transmitancia. Debido a que la potencia incidente de la fuente y la sensibilidad del detector varían con la longitud de onda, el fotómetro se recalibra cada vez que se cambia el filtro. Los fotómetros tienen la ventaja de ser relativamente económicos, resistentes y fáciles de mantener. Otra ventaja de un fotómetro es su portabilidad, lo que hace que sea fácil de llevar al campo. Las desventajas de un fotómetro incluyen la incapacidad de registrar un espectro de absorción y el ancho de banda efectivo relativamente grande de la fuente, lo que limita la linealidad de la curva de calibración.
El porcentaje de transmitancia varía entre 0% y 100%. Como aprendimos de la Figura 10.2.7, utilizamos un blanco para determinar P 0, que corresponde al 100% T. Incluso en ausencia de luz el detector registra una señal. Cerrar el obturador nos permite asignar 0% T a esta señal. Juntos, el ajuste de 0% T y 100% T calibra el instrumento. La cantidad de luz que pasa a través de una muestra produce una señal que es mayor o igual a 0% T y menor o igual a 100% T.

Figura 10.3.1 . Diagrama esquemático de un fotómetro de filtro. El analista o bien inserta un filtro removible o los filtros se colocan en un carrusel, ejemplo del cual se muestra en el recuadro fotográfico. El analista selecciona un filtro girándolo en su lugar.
Espectrofotómetro de haz único. Un instrumento que utiliza un monocromador para la selección de longitud de onda se llama espectrofotómetro. El espectrofotómetro más simple es un instrumento de haz único equipado con un monocromador de longitud de onda fija (Figura 10.3.2 ). Los espectrofotómetros de haz único se calibran y utilizan de la misma manera que un fotómetro. Un ejemplo de un espectrofotómetro de un solo haz es Spectronic 20D+ de Thermo Scientific, que se muestra en el inserto fotográfico de la Figura 10.3.2 . El Spectronic 20D+ tiene un rango de longitud de onda de 340—625 nm (950 nm cuando se usa un detector sensible al rojo) y un ancho de banda efectivo fijo de 20 nm. Hay disponibles espectrofotómetros de un solo haz de mano que funcionan con baterías, que son fáciles de transportar al campo. Otros espectrofotómetros de haz único también están disponibles con anchos de banda efectivos de 2—8 nm. Los espectrofotómetros de haz único de longitud de onda fija no son prácticos para registrar espectros porque ajustar manualmente la longitud de onda y recalibrar el espectrofotómetro es incómodo y requiere mucho tiempo. La precisión de un espectrofotómetro de haz único está limitada por la estabilidad de su fuente y detector a lo largo del tiempo.

Espectrofotómetro de Doble Haz. Las limitaciones de un espectrofotómetro de haz único de longitud de onda fija se minimizan mediante el uso de un espectrofotómetro de doble haz (Figura 10.3.3 ). Un helicóptero controla la trayectoria de la radiación, alternándola entre la muestra, el blanco y un obturador. El procesador de señales utiliza la velocidad de rotación del helicóptero para resolver la señal que llega al detector en la transmisión de la pieza en blanco, P 0, y la muestra, P T. Al incluir una superficie opaca como obturador, también es posible ajustar continuamente 0% T. El ancho de banda efectivo de un espectrofotómetro de doble haz se controla ajustando las ranuras de entrada y salida del monocromador. Los anchos de banda efectivos de 0.2—3.0 nm son comunes. Un monocromador de barrido permite el registro automatizado de espectros. Los instrumentos de doble haz son más versátiles que los instrumentos de haz simple, siendo útiles tanto para análisis cuantitativos como cualitativos, pero también son más caros y no particularmente portátiles.

Espectrómetro de matriz de diodos. Un instrumento con un solo detector puede monitorear solo una longitud de onda a la vez. Si reemplazamos un solo fotomultiplador con una matriz de fotodiodos, podemos usar el detector resultante para registrar un espectro completo en tan solo 0.1 s. En un espectrómetro de matriz de diodos la radiación de origen pasa a través de la muestra y es dispersada por una rejilla (Figura 10.3.4 ). El detector de matriz de fotodiodos está situado en el plano focal de la rejilla, registrando cada diodo la potencia radiante en un rango estrecho de longitudes de onda. Debido a que reemplazamos un monocromador completo con solo una rejilla, un espectrómetro de matriz de diodos es pequeño y compacto.

Una ventaja de un espectrómetro de matriz de diodos es la velocidad de adquisición de datos, lo que nos permite recolectar múltiples espectros para una sola muestra. Se agregan espectros individuales y se promedian para obtener el espectro final. Este promedio de señal mejora la relación señal/ruido de un espectro. Si sumamos n espectros, la suma de la señal en cualquier punto, x, aumenta como nS x, donde S x es la señal. El ruido en cualquier punto, N x, es un evento aleatorio, que aumenta como\(\sqrt{n} N_x\) cuando sumamos n espectros. La relación señal/ruido después de n escaneos, (S/N) n es
\[\left(\frac{S}{N}\right)_{n}=\frac{n S_{x}}{\sqrt{n} N_{x}}=\sqrt{n} \frac{S_{x}}{N_{x}} \nonumber\]
donde S x/N x es la relación señal/ruido para un solo escaneo. El impacto del promedio de la señal se muestra en la Figura 10.3.5 . El primer espectro muestra la señal después de una exploración, que consiste en un único pico ruidoso. El promedio de señal usando 4 escaneos y 16 escaneos disminuye el ruido y mejora la relación señal/ruido. Una desventaja de una matriz de fotodiodos es que el ancho de banda efectivo por diodo es aproximadamente un orden de magnitud mayor que el de un monocromador de alta calidad.

Para obtener más detalles sobre las señales y el ruido, consulte Introducción a las señales y el ruido de Steven Petrovic, un recurso en línea que forma parte de la Biblioteca Digital de Ciencias Analíticas.
Celdas de Muestra. El compartimento de muestras proporciona un ambiente hermético a la luz que limita la radiación parásita. Las muestras normalmente están en estado líquido o en solución, y se colocan en celdas construidas con materiales transparentes UV/Vis, como cuarzo, vidrio y plástico (Figura 10.3.6 ). Se requiere una celda de cuarzo o sílice fundida cuando se trabaja a una longitud de onda <300 nm donde otros materiales muestran una absorción significativa. La longitud de trayectoria más común es de 1 cm (10 mm), aunque hay disponibles celdas con longitudes de trayectoria más cortas (tan solo 0.1 cm) y más largas (hasta 10 cm). Las celdas de mayor longitud de trayectoria son útiles cuando se analiza una solución muy diluida o para muestras de gas. Las celdas de la más alta calidad permiten que la radiación golpee una superficie plana en un ángulo de 90 o, minimizando la pérdida de radiación a la reflexión. Un tubo de ensayo a menudo se usa como celda de muestra con instrumentos simples de un solo haz, aunque las diferencias en la longitud de trayectoria de la celda y las propiedades ópticas agregan una fuente adicional de error al análisis.

Si necesitamos monitorear la concentración de un analito a lo largo del tiempo, es posible que no sea posible extraer muestras para su análisis. Este suele ser el caso, por ejemplo, cuando se monitorea una línea de producción industrial o línea de desechos, cuando se monitorea la sangre de un paciente, o cuando se monitorea un sistema ambiental, como una corriente. Con una sonda de fibra óptica podemos analizar muestras in situ. Un ejemplo de una sonda de fibra óptica de detección remota se muestra en la Figura 10.3.7 . La sonda consta de dos haces de cable de fibra óptica. Un haz transmite radiación desde la fuente a la punta de la sonda, que está diseñada para permitir que la muestra fluya a través de la celda de muestra. La radiación de la fuente pasa a través de la solución y es reflejada de nuevo por un espejo. El segundo haz de cable de fibra óptica transmite la radiación no absorbida al selector de longitud de onda. Otro diseño reemplaza la celda de flujo mostrada en la Figura 10.3.7 con una membrana que contiene un reactivo que reacciona con el analito. Cuando el analito se difunde en la membrana reacciona con el reactivo, produciendo un producto que absorbe radiación UV o visible. La radiación no absorbida de la fuente se refleja o dispersa de nuevo al detector. Las sondas de fibra óptica que muestran selectividad química se denominan optrodos [(a) Seitz, W. R. Anal. Chem. 1984, 56, 16A—34A; b) Angel, S. M. Espectroscopia 1987, 2 (2), 38—48].

Diseños de instrumentos para adsorción por infrarrojos
Fotómetro de Filtro. El instrumento más simple para la espectroscopia de absorción IR es un fotómetro de filtro similar al mostrado en la Figura 10.3.1 para absorción UV/Vis. Estos instrumentos tienen la ventaja de la portabilidad y normalmente se utilizan como analizadores dedicados para gases como HCN y CO.
Espectrofotómetro de doble haz. Los instrumentos infrarrojos que utilizan un monocromador para la selección de longitudes de onda utilizan ópticas de doble haz similar a la que se muestra en la Figura 10.3.3 . Se prefieren las ópticas de doble haz sobre las ópticas de haz único porque las fuentes y los detectores de radiación infrarroja son menos estables que los de la radiación UV/Vis. Además, es más fácil corregir la absorción de la radiación infrarroja por el CO 2 atmosférico y el vapor H 2 O cuando se utilizan ópticas de doble haz. Las resoluciones de 1—3 cm —1 son típicas para la mayoría de los instrumentos.
Espectrómetro de transformada de Fourier. En un espectrómetro infrarrojo de transformada de Fourier, o FT—IR, el monocromador se reemplaza por un interferómetro (Figura 10.1.13). Debido a que un FT-IR incluye solo una única trayectoria óptica, es necesario recolectar un espectro separado para compensar la absorbancia del CO 2 atmosférico y el vapor H 2 O. Esto se hace recogiendo un espectro de fondo sin la muestra y almacenando el resultado en la memoria de la computadora del instrumento. El espectro de fondo se elimina del espectro de la muestra tomando la relación de las dos señales. En comparación con otros diseños de instrumentos, un FT—IR proporciona una rápida adquisición de datos, lo que permite una mejora en la relación señal/ruido a través del promedio de señal.
Celdas de Muestra. La espectroscopia infrarroja se utiliza rutinariamente para analizar muestras de gas, líquidos y sólidos. Las células de muestra están hechas de materiales, como NaCl y KBr, que son transparentes a la radiación infrarroja. Los gases se analizan utilizando una celda con una longitud de trayectoria de aproximadamente 10 cm. Las longitudes de trayectoria más largas se obtienen mediante el uso de espejos para pasar el haz de radiación a través de la muestra varias veces.
Una muestra líquida puede analizarse usando una variedad de células de muestra diferentes (Figura 10.3.8 ). Para líquidos no volátiles se prepara una muestra adecuada colocando una gota del líquido entre dos placas de NaCl, formando una película delgada que típicamente tiene menos de 0.01 mm de espesor. Los líquidos volátiles se colocan en una celda sellada para evitar su evaporación.

El análisis de las muestras de solución está limitado por las propiedades de absorción de IR del solvente, siendo CCl 4, CS 2 y CHCl 3 los solventes más comunes. Las soluciones se colocan en celdas que contienen dos ventanas de NaCl separadas por un espaciador de teflón. Al cambiar el espaciador de teflón, se obtienen longitudes de trayectoria de 0.015—1.0 mm.
Las muestras sólidas transparentes se analizan colocándolas directamente en el haz IR. La mayoría de las muestras sólidas, sin embargo, son opacas y primero se dispersan en un medio más transparente antes de registrar el espectro IR. Si se dispone de un disolvente adecuado, entonces el sólido se analiza preparando una solución y analizando como se describió anteriormente. Cuando no se dispone de un disolvente adecuado, las muestras sólidas se analizan preparando una mezcla de la muestra finamente pulverizada con un aceite adecuado. Alternativamente, la muestra en polvo se mezcla con KBr y se prensa en un pellet ópticamente transparente.
El análisis de una muestra acuosa se complica por la solubilidad de la ventana celular de NaCl en agua. Un enfoque para obtener un espectro infrarrojo de una solución acuosa es usar reflectancia total atenuada en lugar de transmisión. La Figura 10.3.9 muestra un diagrama de un instrumento FT—IR típico de reflectancia total atenuada (ATR). La celda ATR consiste en un material de alto índice de refracción, como ZnSe o diamante, intercalado entre un sustrato de bajo índice de refracción y una muestra de menor índice de refracción. La radiación de la fuente ingresa al cristal ATR donde experimenta una serie de reflexiones internas antes de salir del cristal. Durante cada reflexión la radiación penetra en la muestra a una profundidad de unas pocas micras, lo que resulta en una atenuación selectiva de la radiación en aquellas longitudes de onda donde la muestra absorbe. Los espectros ATR son similares, pero no idénticos, a los obtenidos al medir la transmisión de radiación.
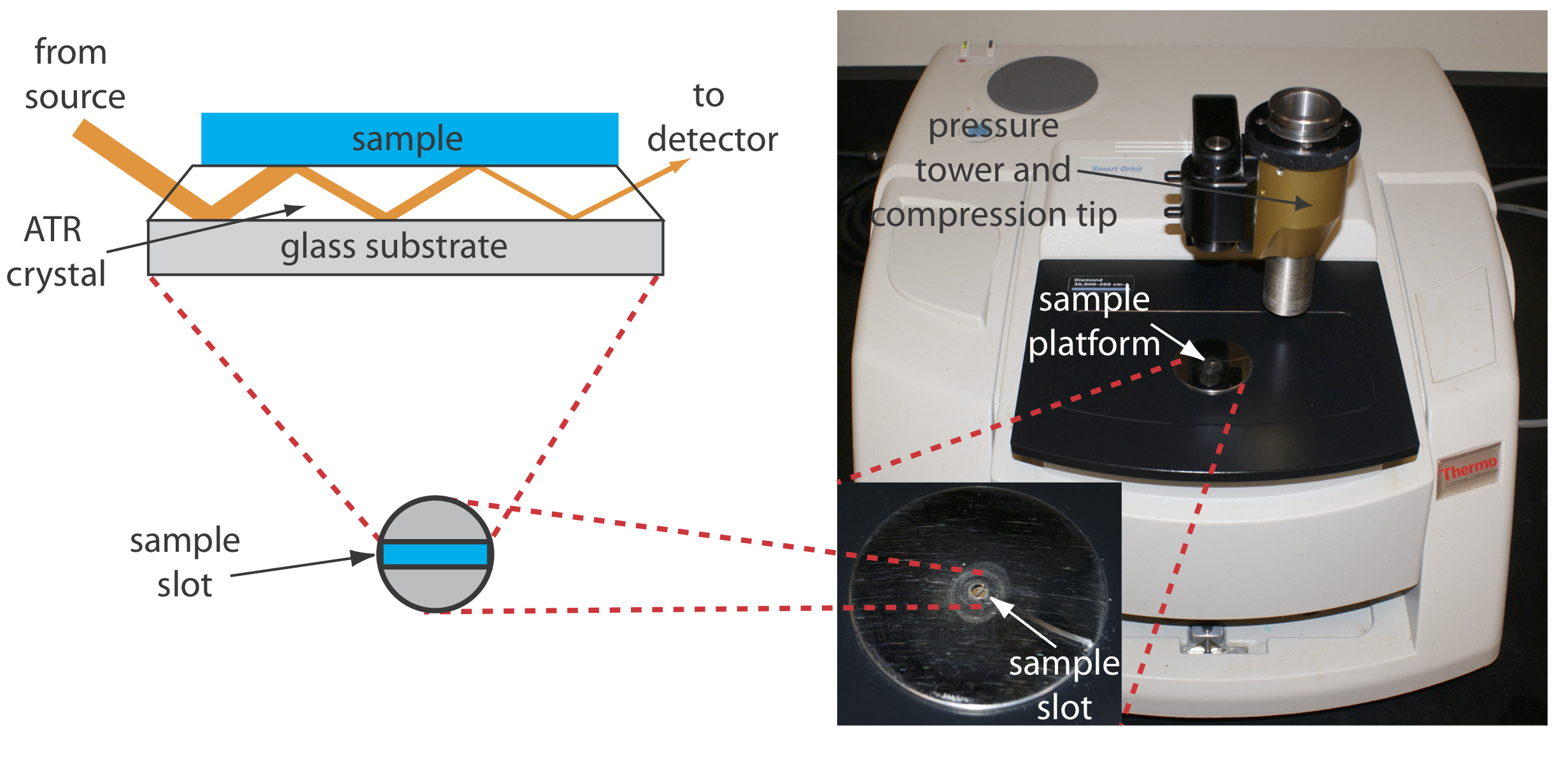
Las muestras sólidas también se pueden analizar usando una célula de muestra ATR. Después de colocar el sólido en la ranura de la muestra, una punta de compresión asegura que esté en contacto con el cristal ATR. Ejemplos de sólidos analizados por ATR incluyen polímeros, fibras, telas, polvos y muestras de tejido biológico. Otro método de reflectancia es la reflectancia difusa, en la que la radiación se refleja desde una superficie rugosa, como un polvo. Las muestras en polvo se mezclan con un material no absorbente, como KBr en polvo, y se recoge y analiza la luz reflejada. Al igual que con ATR, el espectro resultante es similar al obtenido por los métodos de transmisión convencionales. Más detalles sobre estos y otros métodos para preparar sólidos para el análisis infrarrojo se pueden encontrar en los recursos adicionales de este capítulo.
Aplicaciones Cuantitativas
La determinación de la concentración de un analito basada en su absorción de radiación ultravioleta o visible es uno de los métodos analíticos cuantitativos más frecuentes. Una razón de su popularidad es que muchos compuestos orgánicos e inorgánicos tienen fuertes bandas de absorción en la región UV/Vis del espectro electromagnético. Además, si un analito no absorbe radiación UV/Vis, o si su absorbancia es demasiado débil, a menudo podemos reaccionar con otra especie que absorbe fuertemente. Por ejemplo, una solución diluida de Fe 2 + no absorbe la luz visible. Reaccionar Fe 2 + con o -fenantrolina, sin embargo, forma un complejo naranja-rojo\(\text{Fe(phen)}_3^{2+}\) que tiene una banda de absorbancia fuerte y ancha cerca de 500 nm. Una ventaja adicional de la absorción UV/Vis es que en la mayoría de los casos es relativamente fácil ajustar las condiciones experimentales e instrumentales para que se obedezca la ley de Beer.
Un análisis cuantitativo basado en la absorción de radiación infrarroja, aunque importante, se encuentra con menor frecuencia que con la absorción UV/Vis. Una razón es la mayor tendencia a desviaciones instrumentales de la ley de Beer cuando se utiliza radiación infrarroja. Debido a que una banda de absorción infrarroja es relativamente estrecha, cualquier desviación debida a la falta de radiación monocromática es más pronunciada. Además, las fuentes infrarrojas son menos intensas que las fuentes UV/Vis, lo que hace que la radiación parásita sea un problema mayor. Las diferencias entre las longitudes de trayectoria para muestras y para estándares cuando se usan películas líquidas delgadas o pellets de KBr son un problema, aunque un estándar interno puede corregir cualquier diferencia en la longitud de la trayectoria. Finalmente, establecer una línea base 100% T (A = 0) a menudo es difícil debido a que las propiedades ópticas de las células de muestra de NaCl pueden cambiar significativamente con la longitud de onda debido a la contaminación y degradación. Podemos minimizar este problema midiendo la absorbancia relativa a una línea base establecida para la banda de absorción. La figura 10.3.10 muestra cómo se logra esto.

Otro enfoque es usar una celda con una longitud de ruta fija, como la que se muestra en la Figura 10.3.8 b.
Aplicaciones Ambientales
El análisis de aguas y aguas residuales a menudo se basa en la absorción de radiación ultravioleta y visible. Muchos de estos métodos se describen en la Tabla 10.3.1 . Varios de estos métodos se describen aquí con más detalle.
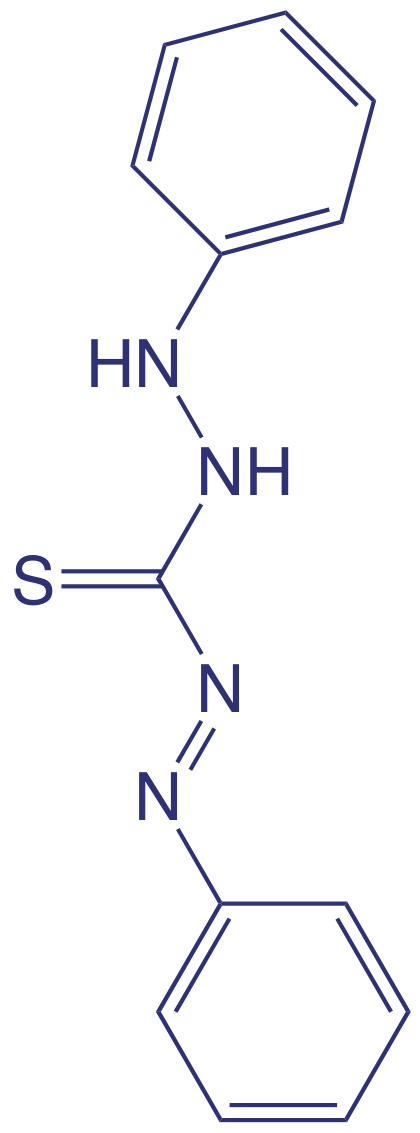
Aunque el análisis cuantitativo de metales en aguas y aguas residuales se realiza principalmente por absorción atómica o espectroscopía de emisión atómica, muchos metales también se pueden analizar después de la formación de un complejo metal-ligando coloreado. Una ventaja de estos métodos espectroscópicos es que se adaptan fácilmente al análisis de muestras en campo usando un fotómetro de filtro. Un ligando utilizado para el análisis de varios metales es la difeniltiocarbazona, también conocida como ditizona. La ditizona no es soluble en agua, pero cuando una solución de ditizona en CHCl 3 se agita con una solución acuosa que contiene un ion metálico apropiado, se forma un complejo metal-ditizonato coloreado que es soluble en CHCl 3. La selectividad de la ditizona se controla ajustando el pH de la muestra. Por ejemplo, Cd 2 + se extrae de soluciones hechas fuertemente básicas con NaOH, Pb 2 + de soluciones hechas básicas con un tampón NH 3/NH 4 +, y Hg 2 + de soluciones que son ligeramente ácidas.
La estructura de la ditizona se muestra a continuación. Consulte el Capítulo 7 para una discusión sobre la extracción de iones metálicos usando ditizona.
Cuando se agrega cloro al agua, la porción disponible para la desinfección se llama cloro residual. Hay dos formas de cloro residual. El residuo de cloro libre incluye Cl 2, HOCl y OCl —. El cloro residual combinado, que se forma a partir de la reacción de NH 3 con HOCl, consiste en monocloramina, NH 2 Cl, dicloramina, NHCl 2 y tricloramina, NCl 3. Debido a que el residuo de cloro libre es más eficiente como desinfectante, existe un interés en métodos que puedan distinguir entre las diferentes formas del residuo de cloro total. Uno de esos métodos es el método del violeta de cristal leuco. El cloro residual libre se determina mediante la adición de violeta de cristal leuco a la muestra, el cual se oxida instantáneamente para dar un compuesto de color azul que se monitorea a 592 nm. Completar el análisis en menos de cinco minutos evita una posible interferencia del cloro residual combinado. El cloro residual total (libre + combinado) se determina haciendo reaccionar una muestra separada con yoduro, el cual reacciona con ambos residuos de cloro para formar HOI. Cuando se completa la reacción, se agrega violeta de cristal leuco y se oxida por HOI, dando el mismo producto de color azul. El cloro residual combinado se determina por diferencia.
En el Capítulo 9 exploramos cómo se puede determinar el residuo de cloro total mediante una valoración redox; ver Método Representativo 9.4.1 para más detalles. El método aquí descrito nos permite dividir el cloro residual total en sus partes componentes.
La concentración de fluoruro en el agua potable está determinada indirectamente por su capacidad para formar un complejo con el circonio. En presencia del colorante SPADNS, una solución de circonio forma un compuesto de color rojo, llamado lago, que absorbe a 570 nm. Cuando se agrega fluoruro, la formación del\(\text{ZrF}_6^{2-}\) complejo estable provoca que una porción del lago se disocie, disminuyendo la absorbancia. Una gráfica de absorbancia versus la concentración de fluoruro, por lo tanto, tiene una pendiente negativa.
SPADNS, cuya estructura se muestra a continuación, es una abreviatura de la sal sódica del ácido 2- (4-sulfofenilazo) -1,8-dihidroxi-3,6-naftalenodisulfónico, que es un bocado para decir.

También se utilizan métodos espectroscópicos para determinar los constituyentes orgánicos en el agua. Por ejemplo, las concentraciones combinadas de fenol y fenoles orto y meta-sustituidos se determinan mediante destilación al vapor para separar los fenoles de las impurezas no volátiles. El destilado reacciona con 4-aminoantipirina a pH 7.9 ± 0.1 en presencia de K 3 Fe (CN) 6 a un tinte antipirina de color amarillo. Después de extraer el colorante en CHCl 3, se monitoriza su absorbancia a 460 nm. Se prepara una curva de calibración utilizando únicamente el fenol no sustituído, C 6 H 5 OH. Debido a que la absortividad molar de los fenoles sustituidos generalmente es menor que la del fenol, la concentración reportada representa la concentración mínima de compuestos fenólicos.
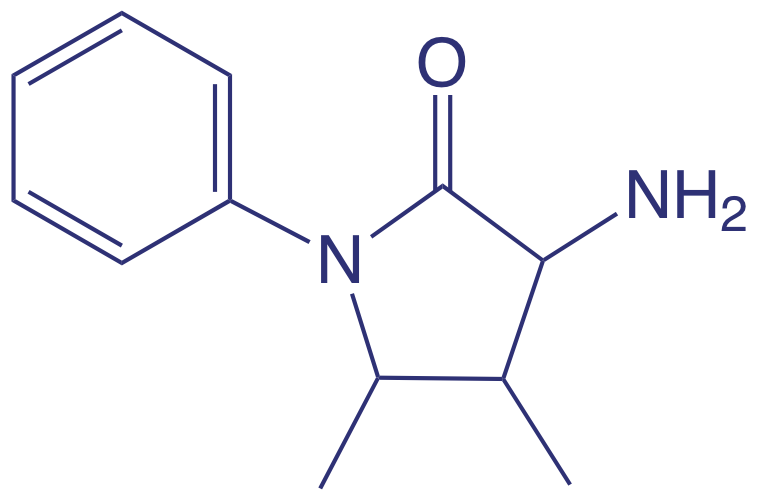
4-aminoantipireno
La absorción molecular también se utiliza para el análisis de contaminantes atmosféricos significativos desde el punto de vista ambiental. En muchos casos el análisis se realiza recogiendo la muestra en agua, convirtiendo el analito en una forma acuosa que puede ser analizada por métodos como los descritos en la Tabla 10.3.1 . Por ejemplo, la concentración de NO 2 se determina oxidando NO 2 a\(\text{NO}_3^-\). La concentración de\(\text{NO}_3^-\) se determina luego reduciéndola primero a\(\text{NO}_2^-\) con Cd, y luego reaccionando\(\text{NO}_2^-\) con sulfanilamida y N - (1-naftil) -etilendiamina para formar un colorante azoico rojo. Otra aplicación importante es el análisis para SO 2, el cual se determina mediante la recolección de la muestra en una solución acuosa de\(\text{HgCl}_4^{2-}\) donde reacciona para formar\(\text{Hg(SO}_3)_2^{2-}\). La adición de p-rosanilina y formaldehído produce un complejo púrpura que se monitorea a 569 nm. La absorción infrarroja es útil para el análisis de vapores orgánicos, incluyendo HCN, SO 2, nitrobenceno, metil mercaptano y cloruro de vinilo. Con frecuencia, estos análisis se realizan utilizando fotómetros infrarrojos portátiles dedicados.
Aplicaciones Clínicas
El análisis de muestras clínicas a menudo se complica por la complejidad de la matriz de la muestra, lo que puede contribuir a una absorción de fondo significativa a la longitud de onda deseada. La determinación de los barbitúricos séricos proporciona un ejemplo de cómo se supera este problema. Los barbitúricos se extraen primero de una muestra de suero con CHCl 3 y luego se extraen del CHCl 3 en NaOH 0.45 M (pH ≈ 13). La absorbancia del extracto acuoso se mide a 260 nm, e incluye contribuciones de los barbitúricos así como otros componentes extraídos de la muestra de suero. Luego se baja el pH de la muestra a aproximadamente 10 mediante la adición de NH 4 Cl y se vuelve a medir la absorbancia. Debido a que los barbitúricos no absorben a este pH, podemos usar la absorbancia a pH 10, A pH 10, para corregir la absorbancia a pH 13, A pH 13
\[A_\text{barb} = A_\text{pH 13} - \frac {V_\text{samp} + V_{\text{NH}_4\text{Cl}}} {V_\text{samp}} \times A_\text{pH 10} \nonumber\]
donde A barb es la absorbancia debida a los barbitúricos séricos y V samp y\(V_{\text{NH}_4\text{Cl}}\) son los volúmenes de muestra y NH 4 Cl, respectivamente. El cuadro 10.3.2 proporciona un resumen de varios otros métodos para analizar muestras clínicas.
Aplicaciones Industriales
La absorción molecular UV/Vis se utiliza para el análisis de una amplia gama de muestras industriales, incluyendo productos farmacéuticos, alimentos, pintura, vidrio y metales. En muchos casos los métodos son similares a los descritos en Table 10.3.1 y en Table 10.3.2 . Por ejemplo, la cantidad de hierro en los alimentos se determina poniendo el hierro en solución y analizándolo utilizando el método o -fenantrolina listado en la Tabla 10.3.1 .
Muchos compuestos farmacéuticos contienen cromóforos que los hacen adecuados para su análisis por absorción UV/Vis. Los productos analizados de esta manera incluyen antibióticos, hormonas, vitaminas y analgésicos. Un ejemplo del uso de la absorción UV es en la determinación de la pureza de las tabletas de aspirina, para lo cual el ingrediente activo es el ácido acetilsalicílico. El ácido salicílico, que se produce por la hidrólisis del ácido acetilsalicílico, es una impureza indeseable en los comprimidos de aspirina, y no debe estar presente a más de 0.01% w/w; las muestras se tamizan para detectar niveles inaceptables de ácido salicílico monitoreando la absorbancia a una longitud de onda de 312 nm. El ácido acetilsalicílico absorbe a 280 nm, pero absorbe mal a 312 nm. Se eligen las condiciones para preparar la muestra de manera que una absorbancia mayor a 0.02 signifique un nivel inaceptable de ácido salicílico.
Aplicaciones Forenses
La absorción molecular UV/Vis se utiliza rutinariamente para el análisis de narcóticos y para pruebas de drogas. Una aplicación forense interesante es la determinación de alcohol en sangre mediante la prueba de alcoholímetro. En esta prueba se burbujea una muestra de aliento de 52.5-mL a través de una solución acidificada de K 2 Cr 2 O 7, que oxida el etanol a ácido acético. La concentración de etanol en la muestra de aliento se determina por una disminución en la absorbancia a 440 nm donde absorbe el ion dicromato. Un contenido de alcohol en sangre de 0.10%, que está por encima del límite legal, corresponde a 0.025 mg de etanol en la muestra de aliento.
Desarrollo de un método cuantitativo para un solo componente
Para desarrollar un método analítico cuantitativo, se deben establecer las condiciones bajo las cuales se obedece la ley de Beer. Primero, la longitud de onda más apropiada para el análisis se determina a partir de un espectro de absorción. En la mayoría de los casos la mejor longitud de onda corresponde a un máximo de absorción porque proporciona mayor sensibilidad y es menos susceptible a limitaciones instrumentales. Segundo, si el instrumento tiene ranuras ajustables, entonces se elige un ancho de hendidura apropiado. El espectro de absorción también ayuda a seleccionar un ancho de hendidura al elegir un ancho que sea lo suficientemente estrecho como para evitar las limites- ciones instrumentales a la ley de Beer, pero lo suficientemente amplio como para aumentar el rendimiento de la radiación de la fuente. Finalmente, se construye una curva de calibración para determinar el rango de concentraciones para el cual es válida la ley de Beer. Consideraciones adicionales que son importantes en cualquier método cuantitativo son el efecto de los potenciales interferentes y el establecimiento de un blanco apropiado.
Método Representativo 10.3.1: Determinación de Hierro en Agua y Aguas Residuales
La mejor manera de apreciar los detalles teóricos y prácticos discutidos en esta sección es examinar cuidadosamente un método analítico típico. Aunque cada método es único, la siguiente descripción de la determinación de hierro en agua y aguas residuales proporciona un ejemplo instructivo de un procedimiento típico. La descripción aquí se basa en el Método 3500-Fe B publicado en Standard Methods for the Examination of Water and Wastewater, 20th Ed., American Public Health Association: Washington, D. C., 1998.
Descripción del método
El hierro en el estado de oxidación +2 reacciona con o -fenantrolina para formar el\(\text{Fe(phen)}_3^{2+}\) complejo naranja-rojo. La intensidad del color del complejo es independiente de la acidez de la solución entre un pH de 3 y 9. Debido a que el complejo se forma más rápidamente a niveles de pH más bajos, la reacción generalmente se lleva a cabo dentro de un rango de pH de 3.0—3.5. Cualquier hierro presente en el estado de oxidación +3 se reduce con hidroxilamina antes de añadir o -fenantrolina. Los interferentes más importantes son agentes oxidantes fuertes, polifosfatos e iones metálicos como Cu 2 +, Zn 2+, Ni 2 + y Cd 2 +. Una interferencia de los agentes oxidantes se minimiza añadiendo un exceso de hidroxilamina, y una interferencia del polifosfato se minimiza al hervir la muestra en presencia de ácido. La absorbancia de muestras y patrones se mide a una longitud de onda de 510 nm usando una celda de 1 cm (también se pueden usar celdas de longitud de ruta más larga). La ley de la cerveza se obedece para concentraciones dentro del rango de 0.2—4.0 mg Fe/L.
Procedimiento
Para una muestra que contenga menos de 2 mg de Fe/L, transfiera directamente una porción de 50 mL a un matraz Erlenmeyer de 125 mL. Las muestras que contienen más de 2 mg de Fe/L se diluyen antes de adquirir la porción de 50 ml. Agregar 2 mL de HCl concentrado y 1 mL de hidroxilamina a la muestra. Llevar la solución a ebullición y continuar hirviendo hasta que el volumen de la solución se reduzca a entre 15 y 20 mL. Después de enfriar a temperatura ambiente, transferir la solución a un matraz aforado de 50 mL, agregar 10 mL de un tampón de acetato amónico, 2 mL de una solución de 1000 ppm de o-fenantrolina, y diluir a volumen. Permita 10—15 minutos para el desarrollo del color antes de medir la absorbancia, usando agua destilada para establecer 100% T. Los estándares de calibración, incluyendo un blanco, se preparan por el mismo procedimiento usando una solución madre que contiene una concentración conocida de Fe 2 +.
Preguntas
1. Explique por qué los agentes oxidantes fuertes son interferentes y por qué un exceso de hidroxilamina evita la interferencia.
Un agente oxidante fuerte oxidará algo de Fe 2 + a Fe 3 +. Debido a\(\text{Fe(phen)}_3^{3+}\) que no absorbe tan fuertemente como\(\text{Fe(phen)}_3^{2+}\), la absorbancia es menor de lo esperado, lo que produce un error determinado negativo. El exceso de hidroxilamina reacciona con los agentes oxidantes, retirándolos de la solución.
2. El color del complejo es estable entre niveles de pH de 3 y 9. ¿Cuáles son algunas posibles complicaciones a pH más ácidos o más básicos?
Debido a que la o-fenantrolina es una base débil, su constante de formación condicional\(\text{Fe(phen)}_3^{2+}\) se vuelve más pequeña a niveles de pH más ácidos, donde la o-fenantrolina está presente en su forma protonada. El resultado es una disminución en la absorbancia y un método analítico menos sensible. Cuando el pH es mayor a 9, la competencia entre OH — y o-fenantrolina por Fe 2 + también disminuye la absorbancia. Además, si el pH es suficientemente básico existe el riesgo de que el hierro precipite como Fe (OH) 2.
3. El cadmio es un interferente porque forma un precipitado con o -fenantrolina. ¿Qué efecto tiene la formación de precipitado en la determinación del hierro?
Debido a que la o-fenantrolina está presente en gran exceso (2000 μg de o-fenantrolina para 100 μg de Fe2 +), no es probable que la interferencia se deba a una cantidad insuficiente de o-fenantrolina disponible para reaccionar con el Fe 2 + . La presencia de un precipitado en la celda muestra da como resultado la dispersión de la radiación, lo que provoca un aumento aparente de la absorbancia. Debido a que la absorbancia medida aumenta, la concentración reportada es demasiado alta. Aunque la dispersión es un problema aquí, puede servir como base de un método analítico útil. Consulte el Capítulo 10.8 para más detalles.
4. Incluso el acetato de amonio de alta calidad contiene una cantidad significativa de hierro. ¿Por qué esta fuente de hierro no es un problema?
Debido a que todas las muestras y patrones se preparan usando el mismo volumen de tampón de acetato de amonio, la contribución de esta fuente de hierro se explica por el blanco de reactivo de la curva de calibración.
Análisis cuantitativo para una sola muestra
Para determinar la concentración de un analito medimos su absorbancia y aplicamos la ley de Beer utilizando cualquiera de los métodos de estandarización descritos en el Capítulo 5. Los métodos más comunes son una curva de calibración normal utilizando estándares externos y el método de adiciones estándar. También es posible una estandarización de un solo punto, aunque primero debemos verificar que la ley de Beer es válida para la concentración de analito en las muestras y el estándar.
La determinación de hierro en una corriente de residuos industriales se realiza mediante la o-fenantrolina descrita en el Método Representativo 10.3.1. Utilizando los datos de la siguiente tabla, determinar los mg Fe/L en el flujo de desechos.
| mg Fe/L | absorbancia |
|---|---|
| 0.00 | 0.000 |
| 1.00 | 0.183 |
| 2.00 | 0.364 |
| 3.00 | 0.546 |
| 4.00 | 0.727 |
| muestra | 0.269 |
Solución
La regresión lineal de la absorbancia frente a la concentración de Fe en los patrones da la curva de calibración y la ecuación de calibración que se muestran aquí

\[A=0.0006+\left(0.1817 \ \mathrm{mg}^{-1} \mathrm{L}\right) \times(\mathrm{mg} \mathrm{Fe} / \mathrm{L}) \nonumber\]
Sustituyendo la absorbancia de la muestra en la ecuación de calibración da la concentración de Fe en la corriente residual como 1.48 mg Fe/L
La concentración de Cu 2 + en una muestra se determina haciéndola reaccionar con el ligando cuprizona y midiendo su absorbancia a 606 nm en una celda de 1.00-cm. Cuando una muestra de 5.00-mL es tratada con cuprizona y diluida a 10.00 mL, la solución resultante tiene una absorbancia de 0.118. Una segunda muestra de 5.00-mL se mezcla con 1.00 mL de un estándar de 20.00 mg/L de Cu 2 +, se trata con cuprizona y se diluye a 10.00 mL, dando una absorbancia de 0.162. Reportan los mg Cu 2 + /L en la muestra.
- Contestar
-
Para esta adición estándar escribimos ecuaciones que relacionan la absorbancia con la concentración de Cu 2 + en la muestra antes de la adición estándar
\[0.118=\varepsilon b \left[ C_{\mathrm{Cu}} \times \frac{5.00 \text{ mL}}{10.00 \text{ mL}}\right] \nonumber\]
y después de la adición estándar
\[0.162=\varepsilon b\left(C_{\mathrm{Cu}} \times \frac{5.00 \text{ mL}}{10.00 \text{ mL}}+\frac{20.00 \ \mathrm{mg} \ \mathrm{Cu}}{\mathrm{L}} \times \frac{1.00 \ \mathrm{mL}}{10.00 \ \mathrm{mL}}\right) \nonumber\]
en cada caso contabilizando la dilución de la muestra original y del estándar. El valor de\(\varepsilon b\) es el mismo en ambas ecuaciones. Resolver cada ecuación para\(\varepsilon b\) e igualar
\[\frac{0.162}{C_{\mathrm{Cu}} \times \frac{5.00 \text{ mL}}{10.00 \text{ mL}}+\frac{20.00 \ \mathrm{mg} \ \mathrm{Cu}}{\mathrm{L}} \times \frac{1.00 \ \mathrm{mL}}{10.00 \ \mathrm{mL}}}=\frac{0.118}{C_{\mathrm{Cu}} \times \frac{5.00 \text{ mL}}{10.00 \text{ mL}}} \nonumber\]
nos deja con una ecuación en la que C Cu es la única variable. Resolviendo para C Cu da su valor como
\[\frac{0.162}{0.500 \times C_{\mathrm{Cu}}+2.00 \ \mathrm{mg} \ \mathrm{Cu} / \mathrm{L}}=\frac{0.118}{0.500 \times C_{\mathrm{Cu}}} \nonumber\]
\[0.0810 \times C_{\mathrm{Cu}}=0.0590 \times C_{\mathrm{Ca}}+0.236 \ \mathrm{mg} \ \mathrm{Cu} / \mathrm{L} \nonumber\]
\[0.0220 \times C_{\mathrm{Cu}}=0.236 \ \mathrm{mg} \ \mathrm{Cu} / \mathrm{L} \nonumber\]
\[C_{\mathrm{Cu}}=10.7 \ \mathrm{mg} \ \mathrm{Cu} / \mathrm{L} \nonumber\]
Análisis Cuantitativo de Mezclas
Supongamos que necesitamos determinar la concentración de dos analitos, X e Y, en una muestra. Si cada analito tiene una longitud de onda donde el otro analito no absorbe, entonces podemos proceder usando el enfoque del Ejemplo 10.3.5 . Desafortunadamente, las bandas de absorción UV/Vis son tan amplias que frecuentemente no es posible encontrar longitudes de onda adecuadas. Debido a que la ley de Beer es aditiva la absorbancia de la mezcla, Una mezcla, es
donde\(\lambda_1\) es la longitud de onda a la que medimos la absorbancia. Debido a que la Ecuación\ ref {10.1} incluye términos para la concentración tanto de X como de Y, la absorbancia a una longitud de onda no proporciona suficiente información para determinar C X o C Y. Si medimos la absorbancia a una segunda longitud de onda
entonces podemos determinar C X y C Y resolviendo simultáneamente la Ecuación\ ref {10.1} y la Ecuación\ ref {10.2}. Por supuesto, también debemos determinar el valor para\(\varepsilon_X\) y\(\varepsilon_Y\) en cada longitud de onda. Para una mezcla de n componentes, debemos medir la absorbancia a n longitudes de onda diferentes.
Las concentraciones de Fe 3 + y Cu 2 + en una mezcla se determinan siguiendo su reacción con hexacianorutenato (II)\(\text{Ru(CN)}_6^{4-}\), que forma un complejo púrpura-azul con Fe 3 + (\(\lambda_\text{max}\)= 550 nm) y un complejo verde pálido con Cu 2 + (\(\lambda_\text{max}\)= 396 nm) [DiTUSA, M. R.; Schlit, A. A. J. Chem. Educ. 1985, 62, 541—542]. Las absorbilidades molares (M —1 cm —1) para los complejos metálicos en las dos longitudes de onda se resumen en la siguiente tabla.
| analito | \(\varepsilon_{550}\) | \(\varepsilon_{396}\) |
|---|---|---|
| Fe 3+ | \ (\ varepsilon_ {550}\) ">9970 | \ (\ varepsilon_ {396}\) ">84 |
| Cu 2+ | \ (\ varepsilon_ {550}\) ">34 | \ (\ varepsilon_ {396}\) ">856 |
Cuando se analiza una muestra que contiene Fe 3 + y Cu 2 + en una celda con una longitud de trayectoria de 1.00 cm, la absorbancia a 550 nm es 0.183 y la absorbancia a 396 nm es 0.109. ¿Cuáles son las concentraciones molares de Fe 3 + y Cu 2 + en la muestra?
Solución
Sustituir valores conocidos en Ecuación\ ref {10.1} y Ecuación\ ref {10.2} da
\[\begin{aligned} A_{550} &=0.183=9970 C_{\mathrm{Fe}}+34 C_{\mathrm{Cu}} \\ A_{396} &=0.109=84 C_{\mathrm{Fe}}+856 C_{\mathrm{Cu}} \end{aligned} \nonumber\]
Para determinar C Fe y C Cu resolvemos la primera ecuación para C Cu
\[C_{\mathrm{Cu}}=\frac{0.183-9970 C_{\mathrm{Fe}}}{34} \nonumber\]
y sustituir el resultado en la segunda ecuación.
\[\begin{aligned} 0.109 &=84 C_{\mathrm{Fe}}+856 \times \frac{0.183-9970 C_{\mathrm{Fe}}}{34} \\ &=4.607-\left(2.51 \times 10^{5}\right) C_{\mathrm{Fe}} \end{aligned} \nonumber\]
Resolviendo para C Fe da la concentración de Fe 3 + como\(1.8 \times 10^{-5}\) M. Sustituyendo esta concentración de nuevo en la ecuación por la absorbancia de la mezcla a 396 nm da la concentración de Cu 2 + como\(1.3 \times 10^{-4}\) M.
Otro enfoque para resolver Ejemplo 10.3.2 es multiplicar la primera ecuación por 856/34 dando
\[4.607=251009 C_{\mathrm{Fe}}+856 C_\mathrm{Cu} \nonumber\]
Restar la segunda ecuación de esta ecuación
\[\begin{aligned} 4.607 &=251009 C_{\mathrm{Fe}}+856 C_{\mathrm{Cu}} \\-0.109 &=84 C_{\mathrm{Fe}}+856 C_{\mathrm{Cu}} \end{aligned} \nonumber\]
da
\[4.498=250925 C_{\mathrm{Fe}} \nonumber\]
y nos encontramos con que C Fe es\(1.8 \times 10^{-5}\). Habiendo determinado C Fe podemos sustituir de nuevo en una de las otras ecuaciones para resolver por C Cu, que es\(1.3 \times 10^{-5}\).
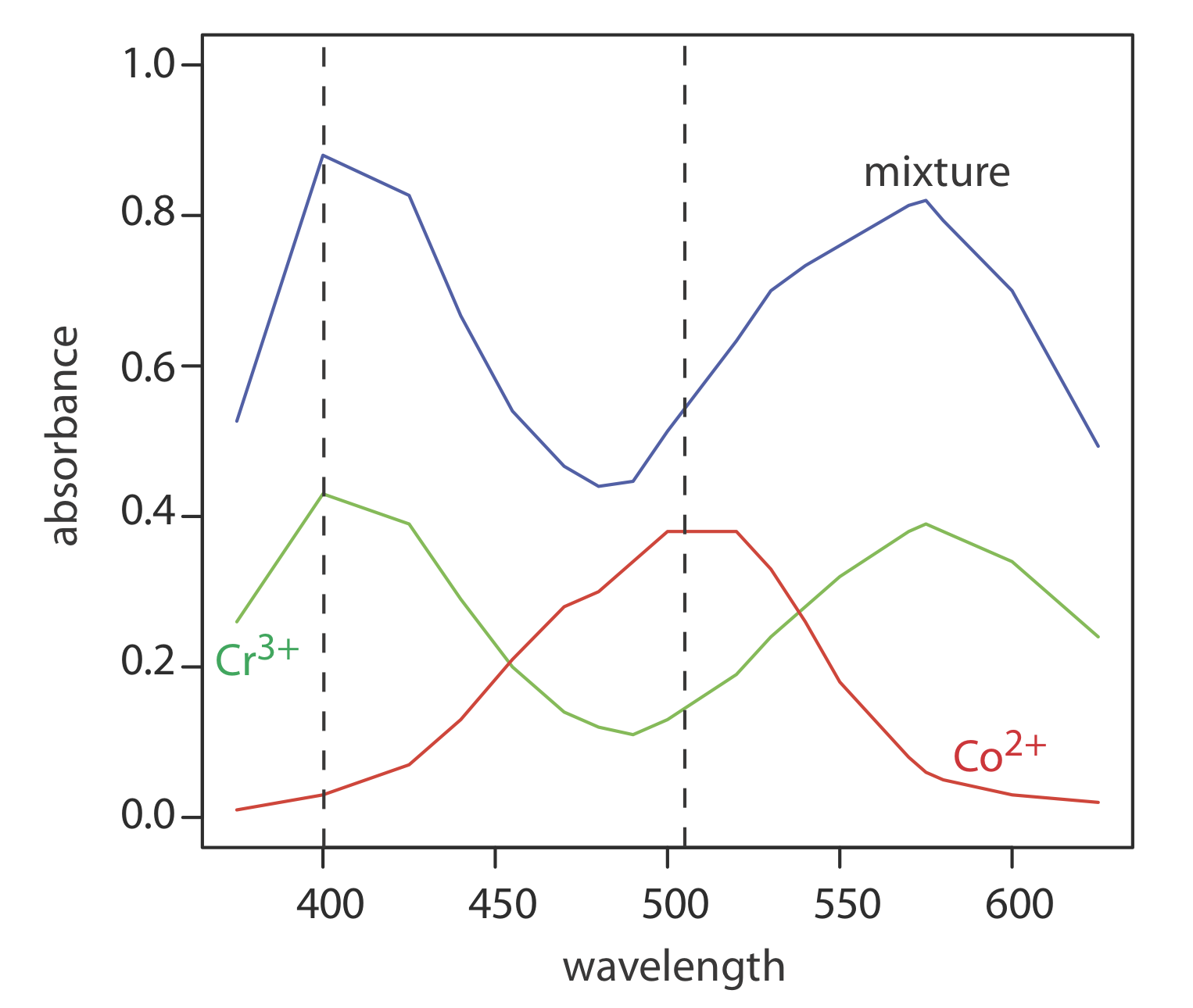
Los espectros de absorbancia para Cr 3 + y Co 2 + se superponen significativamente. Para determinar la concentración de estos analitos en una mezcla, se mide su absorbancia a 400 nm y a 505 nm, produciendo valores de 0.336 y 0.187, respectivamente. Las absorbilidades molares individuales (M —1 cm —1) para Cr3 + son 15.2 a 400 nm y 0.533 a 505 nm; los valores para Co 2 + son 5.60 a 400 nm y 5.07 a 505 nm.
- Contestar
-
Sustituyendo en Ecuación\ ref {10.1} y Ecuación\ ref {10.2} da
\[A_{400} = 0.336 = 15.2C_\text{Cr} + 5.60C_\text{Co} \nonumber\]
\[A_{400} = 0187 = 0.533C_\text{Cr} + 5.07C_\text{Co} \nonumber\]
Para determinar C Cr y C Co resolvemos la primera ecuación para C Co
\[C_{\mathrm{Co}}=\frac{0.336-15.2 \mathrm{C}_{\mathrm{Co}}}{5.60} \nonumber\]
y sustituir el resultado en la segunda ecuación.
\[0.187=0.533 C_{\mathrm{Cr}}+5.07 \times \frac{0.336-15.2 C_{\mathrm{Co}}}{5.60} \nonumber\]
\[0.187=0.3042-13.23 C_{\mathrm{Cr}} \nonumber\]
Resolviendo para C Cr da la concentración de Cr 3 + como\(8.86 \times 10^{-3}\) M. Sustituyendo esta concentración de nuevo en la ecuación por la absorbancia de la mezcla a 400 nm da la concentración de Co 2 + como\(3.60 \times 10^{-2}\) M.
Para obtener resultados con buena precisión y precisión se deben seleccionar las dos longitudes de onda de manera que\(\varepsilon_X > \varepsilon_Y\) a una longitud de onda y\(\varepsilon_X < \varepsilon_Y\) a la otra longitud de onda. Es fácil apreciar por qué esto es cierto. Debido a que la absorbancia en cada longitud de onda está dominada por un analito, cualquier incertidumbre en la concentración del otro analito tiene menor impacto. La Figura 10.3.11 muestra que la elección de longitudes de onda para el Ejercicio de Práctica 10.3.2 es razonable. Cuando la elección de longitudes de onda no es obvia, un método para localizar las longitudes de onda óptimas es graficar\(\varepsilon_X / \varepsilon_y\) como función de la longitud de onda, y determinar las longitudes de onda donde\(\varepsilon_X / \varepsilon_y\) alcanza los valores máximos y mínimos [Mehra, M. C.; Rioux, J. J. Chem. Educ. 1982, 59, 688—689].
Cuando los espectros del analito se superponen severamente, de tal manera que\(\varepsilon_X \approx \varepsilon_Y\) en todas las longitudes de onda, otros métodos computacionales pueden proporcionar una mejor precisión y precisión. En un análisis de regresión lineal multilongitud de onda, por ejemplo, se compara la absorbancia de una mezcla con la de un conjunto de soluciones estándar a varias longitudes de onda [Blanco, M.; Iturriaga, H.; Maspoch, S.; Tarin, P. J. Chem. Educ. 1989, 66, 178—180]. Si A SX y A SY son los valores de absorbancia para soluciones estándar de los componentes X e Y en cualquier longitud de onda, entonces
\[A_{SX}=\varepsilon_{X} b C_{SX} \label{10.3}\]
\[A_{SY}=\varepsilon_{Y} b C_{SY} \label{10.4}\]
donde C SX y C SY son las concentraciones conocidas de X e Y en las soluciones estándar. Resolviendo la ecuación\ ref {10.3} y la ecuación\ ref {10.4} para\(\varepsilon_X\) y para\(\varepsilon_Y\), sustituyendo en la ecuación\ ref {10.1}, y reordenando, da
\[\frac{A_{\operatorname{mix}}}{A_{S X}}=\frac{C_{X}}{C_{S X}}+\frac{C_{Y}}{C_{S Y}} \times \frac{A_{S Y}}{A_{S X}} \nonumber\]
Para determinar C X y C Y, la absorbancia de la mezcla y las absorbancias de las soluciones estándar se miden a varias longitudes de onda. Graficando Una mezcla/A SX versus A SY/A SX da una línea recta con una pendiente de C Y/ C SY y una intersección y de C X/C SX. Este enfoque es particularmente útil cuando no es posible encontrar longitudes de onda donde\(\varepsilon_X > \varepsilon_Y\) y\(\varepsilon_X < \varepsilon_Y\).
El enfoque descrito aquí para una regresión lineal multilongitud de onda utiliza una única solución estándar para cada analito. Un enfoque más riguroso utiliza múltiples estándares para cada analito. La matemática detrás del análisis de tales datos —que llamamos regresión lineal múltiple— está más allá del nivel de este texto. Para más detalles sobre regresión lineal múltiple ver Brereton, R. G. Chemometrics: Data Analysis for the Laboratory and Chemical Plant, Wiley: Chichester, Inglaterra, 2003.
La Figura\(PageIndex{10.11}\) muestra espectros de absorbancia visibles para una solución estándar de 0.0250 M Cr 3 +, una solución estándar de 0.0750 M Co 2 +, y una mezcla que contiene concentraciones desconocidas de cada ion. Aquí se muestran los datos de estos espectros.
| \(\lambda\)(nm) | A Cr | A Cu | Una mezcla | \(\lambda\)(nm) | A Cr | A Cu | Una mezcla |
|---|---|---|---|---|---|---|---|
| \ (\ lambda\) (nm) ">375 | 0.26 | 0.01 | 0.53 | \ (\ lambda\) (nm) ">520 | 0.19 | 0.38 | 0.63 |
| \ (\ lambda\) (nm) ">400 | 0.43 | 0.03 | 0.88 | \ (\ lambda\) (nm) ">530 | 0.24 | 0.33 | 0.70 |
| \ (\ lambda\) (nm) ">425 | 0.39 | 0.07 | 0.83 | \ (\ lambda\) (nm) ">540 | 0.28 | 0.26 | 0.73 |
| \ (\ lambda\) (nm) ">440 | 0.29 | 0.13 | 0.67 | \ (\ lambda\) (nm) ">550 | 0.32 | 0.18 | 0.76 |
| \ (\ lambda\) (nm) ">455 | 0.20 | 0.21 | 0.54 | \ (\ lambda\) (nm) ">570 | 0.38 | 0.08 | 0.81 |
| \ (\ lambda\) (nm) ">470 | 0.14 | 0.28 | 0.47 | \ (\ lambda\) (nm) ">575 | 0.39 | 0.06 | 0.82 |
| \ (\ lambda\) (nm) ">480 | 0.12 | 0.30 | 0.44 | \ (\ lambda\) (nm) ">580 | 0.38 | 0.05 | 0.79 |
| \ (\ lambda\) (nm) ">490 | 0.11 | 0.34 | 0.45 | \ (\ lambda\) (nm) ">600 | 0.34 | 0.03 | 0.70 |
| \ (\ lambda\) (nm) ">500 | 0.13 | 0.38 | 0.51 | \ (\ lambda\) (nm) ">625 | 0.24 | 0.02 | 0.49 |
Utilizar un análisis de regresión multilongitud de onda para determinar la composición de lo desconocido.
Solución
Primero necesitamos calcular los valores para A mix/A SX y para A SY/A SX. Definamos X como Co 2 + e Y como Cr 3 +. Por ejemplo, a una longitud de onda de 375 nm Una mezcla/A SX es 0.53/0.01, o 53 y A SY/A SX es 0.26/0.01, o 26. Completar el cálculo para todas las longitudes de onda y graficar una mezcla/A SX versus A SY/A SX da la curva de calibración mostrada en la Figura 10.3.12 . Ajustar una línea recta a los datos da un modelo de regresión de
\[\frac{A_{\operatorname{mix}}}{A_{S X}}=0.636+2.01 \times \frac{A_{S Y}}{A_{S X}} \nonumber\]
Usando la intersección y, la concentración de Co 2 + es
\[\frac{C_{X}}{C_{S X}}=\frac{\left[\mathrm{Co}^{2+}\right]}{0.0750 \mathrm{M}}=0.636 \nonumber\]
o [Co 2 +] = 0.048 M; usando la pendiente la concentración de Cr 3 + es
\[\frac{C_{Y}}{C_{S Y}}=\frac{\left[\mathrm{Cr}^{3+}\right]}{0.0250 \mathrm{M}}=2.01 \nonumber\]
o [Cr 3 +] = 0.050 M.

Una mezcla de\(\text{MnO}_4^{-}\) y\(\text{Cr}_2\text{O}_7^{2-}\), y estándares de 0.10 mM KMnO 4 y de 0.10 mM K 2 Cr 2 O 7 dan los resultados que se muestran en la siguiente tabla. Determinar la composición de la mezcla. Los datos para este problema son de Blanco, M. C.; Iturriaga, H.; Maspoch, S.; Tarin, P. J. Chem. Educ. 1989, 66, 178—180.
| \(\lambda\)(nm) | A Mn | A Cr | Una mezcla |
|---|---|---|---|
| \ (\ lambda\) (nm) ">266 | 0.042 | 0.410 | 0.766 |
| \ (\ lambda\) (nm) ">288 | 0.082 | 0.283 | 0.571 |
| \ (\ lambda\) (nm) ">320 | 0.168 | 0.158 | 0.422 |
| \ (\ lambda\) (nm) ">350 | 0.125 | 0.318 | 0.672 |
| \ (\ lambda\) (nm) ">360 | 0.036 | 0.181 | 0.366 |
- Contestar
-
Dejando que X represente\(\text{MnO}_4^{-}\) y dejando que Y represente\(\text{Cr}_2\text{O}_7^{2-}\), trazamos la ecuación
\[\frac{A_{\operatorname{mix}}}{A_{SX}}=\frac{C_{X}}{C_{SX}}+\frac{C_{Y}}{C_{S Y}} \times \frac{A_{S Y}}{A_{SX}} \nonumber\]
colocando una mezcla/A SX en el eje y y A SY/A SX en el eje x. Por ejemplo, a una longitud de onda de 266 nm el valor A mix/A SX de es 0.766/0.042, o 18.2, y el valor de A SY/A SX es 0.410 /0.042, o 9.76. Completar los cálculos para todas las longitudes de onda y trazar los datos da el resultado que se muestra aquí

Ajustar una línea recta a los datos da un modelo de regresión de
\[\frac{A_{\text { mix }}}{A_{\text { SX }}}=0.8147+1.7839 \times \frac{A_{SY}}{A_{SX}} \nonumber\]
Usando la intersección y, la concentración de\(\text{MnO}_4^{-}\) es
\[\frac{C_{X}}{C_{S X}}=0.8147=\frac{\left[\mathrm{MnO}_{4}^{-}\right]}{1.0 \times 10^{-4} \ \mathrm{M} \ \mathrm{MnO}_{4}^{-}} \nonumber\]
o\(8.15 \times 10^{-5}\) M\(\text{MnO}_4^{-}\), y usando la pendiente, la concentración de\(\text{Cr}_2\text{O}_7^{2-}\) es
\[\frac{C_{Y}}{C_{S Y}}=1.7839=\frac{\left[\mathrm{Cr}_{2} \mathrm{O}_{7}^{2-}\right]}{1.00 \times 10^{-4} \ \mathrm{M} \ \text{Cr}_{2} \mathrm{O}_{7}^{2-}} \nonumber\]
o\(1.78 \times 10^{-4}\)\(\text{Cr}_2\text{O}_7^{2-}\) M.
Aplicaciones Cualitativas
Como se discute en el Capítulo 10.2, las bandas de absorción ultravioleta, visible e infrarroja resultan de la absorción de radiación electromagnética por electrones o enlaces de valencia específicos. La energía a la que se produce la absorción, y la intensidad de esa absorción, está determinada por el ambiente químico del resto absorbente. Por ejemplo, el benceno tiene varias bandas de absorción ultravioleta debido a\(\pi \rightarrow \pi^*\) las transiciones. La posición e intensidad de dos de estas bandas, 203.5 nm (\(\varepsilon\)= 7400 M —1 cm —1) y 254 nm (\(\varepsilon\)= 204 M —1 cm —1), son sensibles a la sustitución. Para el ácido benzoico, en el que un grupo ácido carboxílico reemplaza a uno de los hidrógenos aromáticos, las dos bandas se desplazan a 230 nm (\(\varepsilon\)= 11600 M —1 cm —1) y 273 nm (\(\varepsilon\)= 970 M —1 cm —1). Se han desarrollado una variedad de reglas para ayudar a correlacionar las bandas de absorción UV/Vis con la estructura química. Correlaciones similares están disponibles para bandas de absorción infrarroja. Por ejemplo, el tramo C=O de un carbonilo es sensible a grupos funcionales adyacentes, apareciendo a 1650 cm —1 para los ácidos, 1700 cm —1 para las cetonas y 1800 cm —1 para los cloruros de ácido. La interpretación de los espectros UV/ Vis e IR recibe una cobertura adecuada en otras partes del plan de estudios de química, especialmente en química orgánica, y no se considera más en este texto.
Con la disponibilidad de adquisición y almacenamiento de datos computarizados es posible construir bibliotecas digitales de espectros de referencia estándar. La identidad de un compuesto desconocido a menudo se puede determinar comparando su espectro con una biblioteca de espectros de referencia, un proceso conocido como búsqueda espectral. Las comparaciones se realizan utilizando un algoritmo que calcula la diferencia acumulativa entre el espectro de la muestra y un espectro de referencia. Por ejemplo, un algoritmo simple usa la siguiente ecuación
\[D = \sum_{i = 1}^n | (A_{sample})_i - (A_{reference})_i | \nonumber\]
donde D es la diferencia acumulativa, Una muestra es la absorbancia de la muestra a longitud de onda o número de onda i, Una referencia es la absorbancia del compuesto de referencia al mismo longitud de onda o número de onda, y n es el número de puntos digitalizados en los espectros. La diferencia acumulada se calcula para cada espectro de referencia. El compuesto de referencia con el valor más pequeño de D es la coincidencia más cercana al compuesto desconocido. La precisión de la búsqueda espectral está limitada por el número y tipo de compuestos incluidos en la biblioteca, y por el efecto de la matriz de la muestra en el espectro.
Otra ventaja de la adquisición computarizada de datos es la capacidad de restar un espectro de otro. Cuando se combina con la búsqueda espectral es posible determinar la identidad de varios componentes en una muestra sin la necesidad de una etapa de separación previa mediante la búsqueda repetida y subtracting de espectros de referencia. Un ejemplo se muestra en la Figura 10.3.13 en la que la composición de una mezcla de dos componentes se determina mediante búsquedas y restas sucesivas. La figura 10.3.13 a muestra el espectro de la mezcla. Una búsqueda en la biblioteca espectral selecciona la cocaina•HCl (Figura 10.3.13 b) como un componente probable de la mezcla. Restar el espectro de referencia para la cocaina•HCl del espectro de la mezcla deja un resultado (Figura 10.3.13 c) que coincide estrechamente con el espectro de referencia del manitol (Figura 10.3.13 d). Restar el espectro de referencia para el manitol deja una pequeña señal residual (Figura 10.3.13 e).

Aplicaciones de Caracterización
La absorción molecular, particularmente en el rango UV/Vis, se ha utilizado para diversos estudios de caracterización, incluyendo la determinación de la estequiometría de complejos metal-ligando y la determinación de constantes de equilibrio. Ambos ejemplos se examinan en esta sección.
Estequiometría de un Complejo Metal-Ligando
Podemos determinar la estequiometría de la reacción de complejación metal-ligando
\[\mathrm{M}+y \mathrm{L} \rightleftharpoons \mathrm{ML}_{y} \nonumber\]
utilizando uno de tres métodos: el método de variaciones continuas, el método de relación molar y el método de relación pendiente. De estos enfoques, el método de variaciones continuas, también llamado método de Job, es el más popular. En este método se prepara una serie de soluciones de tal manera que el total de moles de metal y de ligando, n total, en cada solución es el mismo. Si (n M) i y (n L) i son, respectivamente, los moles de metal y ligando en la solución i, entonces
\[n_{\text { total }}=\ \left(n_{\mathrm{M}}\right)_{i} \ + \ \left(n_{\mathrm{L}}\right)_{i} \nonumber\]
La cantidad relativa de ligando y metal en cada solución se expresa como la fracción molar del ligando, (X L) i, y la fracción molar de metal, (X M) i,
\[\left(X_{\mathrm{L}}\right)_{i}=\frac{\left(n_{\mathrm{L}}\right)_{i}}{n_{\mathrm{total}}} \nonumber\]
\[\left(X_{M}\right)_{i}=1-\frac{\left(n_\text{L}\right)_{i}}{n_{\text { total }}}=\frac{\left(n_\text{M}\right)_{i}}{n_{\text { total }}} \nonumber\]
La concentración del complejo metal-ligando en cualquier solución está determinada por el reactivo limitante, y la mayor concentración ocurre cuando el metal y el ligando se mezclan estequiométricamente. Si monitoreamos la reacción de complejación a una longitud de onda donde solo absorbe el complejo metal-ligando, un gráfico de absorbancia frente a la fracción molar del ligando tiene dos ramificaciones lineales, una cuando el ligando es el reactivo limitante y una segunda cuando el metal es el reactivo limitante. La intersección de las dos ramas representa una mezcla estequiométrica del metal y el ligando. Utilizamos la fracción molar de ligando en la intersección para determinar el valor de y para el complejo metal-ligando ML y.
\[y=\frac{n_{\mathrm{L}}}{n_{\mathrm{M}}}=\frac{X_{\mathrm{L}}}{X_{\mathrm{M}}}=\frac{X_{\mathrm{L}}}{1-X_{\mathrm{L}}} \nonumber\]
También puede graficar los datos como absorbancia versus la fracción molar de metal. En este caso, y es igual a (1 — X M)/X M.
Para determinar la fórmula para el complejo entre Fe 2 + y o-fenantrolina, se prepara una serie de soluciones en las que la concentración total de metal y ligando se mantiene constante a\(3.15 \times 10^{-4}\) M. La absorbancia de cada solución se mide a una longitud de onda de 510 nm. Usando los siguientes datos, determine la fórmula para el complejo.
| X L | absorbancia | X L | absorbancia |
|---|---|---|---|
| 0.000 | 0.000 | 0.600 | 0.693 |
| 0.100 | 0.116 | 0.700 | 0.809 |
| 0.200 | 0.231 | 0.800 | 0.693 |
| 0.300 | 0.347 | 0.900 | 0.347 |
| 0.400 | 0.462 | 1.000 | 0.000 |
| 0.500 | 0.578 |
Solución
En la Figura {{template.index (ID:14)} se muestra una gráfica de absorbancia frente a la fracción molar del ligando. Para encontrar la absorbancia máxima, extrapolamos las dos porciones lineales de la parcela. Las dos líneas se cruzan en una fracción molar de ligando de 0.75. Resolviendo para y da
\[y=\frac{X_{L}}{1-X_{L}}=\frac{0.75}{1-0.75}=3 \nonumber\]
La fórmula para el complejo metal-ligando es\(\text{Fe(phen)}_3^{2+}\).

Utilice los datos de variaciones continuas en la siguiente tabla para determinar la fórmula para el complejo entre Fe 2 + y SCN —. Los datos para este problema se adaptan de Meloun, M.; Havel, J.; Högfeldt, E. Computation of Solution Equilibria, Ellis Horwood: Chichester, Inglaterra, 1988, p. 236.
| X L | absorbancia | X L | absorbancia | X L | absorbancia | X L | absorbancia |
|---|---|---|---|---|---|---|---|
| 0.0200 | 0.068 | 0.2951 | 0.670 | 0.5811 | 0.790 | 0.8923 | 0.324 |
| 0.0870 | 0.262 | 0.3887 | 0.767 | 0.6860 | 0.701 | 0.9787 | 0.071 |
| 0.1792 | 0.471 | 0.4964 | 0.807 | 0.7885 | 0.540 |
- Contestar
-
La siguiente figura muestra una gráfica de variaciones continuas para los datos de este ejercicio. Aunque los puntos de datos individuales muestran una curvatura sustancial, curvatura suficiente que hay poco sentido en tratar de dibujar ramas lineales para exceso de metal y ligando en exceso, la absorbancia máxima ocurre claramente en X L ≈ 0.5. La estequiometría del complejo, por lo tanto, es Fe (SCN) 2+.

Varias precauciones son necesarias al usar el método de variaciones continuas. Primero, el metal y el ligando deben formar solo un complejo metal-ligando. Para determinar si esta condición es verdadera, se construyen gráficas de absorbancia versus X L a varias longitudes de onda diferentes y para varios valores diferentes de n total. Si la absorbancia máxima no ocurre al mismo valor de X L para cada conjunto de condiciones, entonces hay más de un complejo metal-ligando presente. Una segunda precaución es que la absorbancia del complejo metal-ligando debe obedecer la ley de Beer. En tercer lugar, si la constante de formación del complejo metal-ligando es relativamente pequeña, una gráfica de absorbancia versus X L puede mostrar una curvatura significativa. En este caso muchas veces es difícil determinar la estequiometría por extrapolación. Finalmente, debido a que la estabilidad de un complejo metal-ligando puede estar influenciada por las condiciones de la solución, es necesario controlar cuidadosamente la composición de las soluciones. Cuando el ligando es una base débil, por ejemplo, cada solución debe ser tamponada al mismo pH.
En el método de relación molar los moles de un reactivo, generalmente el metal, se mantienen constantes, mientras que los moles del otro reactivo se varían. La absorbancia se monitorea a una longitud de onda donde absorbe el complejo metal-ligando. Una gráfica de absorbancia en función de la relación molar ligando-metal, n L/n M, tiene dos ramas lineales que se cruzan en una relación molar correspondiente a la fórmula del complejo. La Figura 10.3.15 a muestra una gráfica de relación molar para la formación de un complejo 1:1 en el que se monitoriza la absorbancia a una longitud de onda donde solo absorbe el complejo. La Figura 10.3.15 b muestra una gráfica de relación molar para un complejo 1:2 en el que las tres especies, el metal, el ligando y el complejo, absorben a la longitud de onda seleccionada. A diferencia del método de variaciones continuas, el método de relación molar puede usarse para reacciones de complejación que ocurren de manera escalonada si hay una diferencia en las absorbilidades molares de los complejos metal-ligando, y si las constantes de formación son suficientemente diferentes. En la Figura 10.3.15 c. se muestra una gráfica típica de relación molar para la formación escalonada de ML y ML 2.

Tanto para el método de variaciones continuas como para el método de relación molar, determinamos la estequiometría del complejo extrapolando datos de absorbancia a partir de condiciones en las que existe una relación lineal entre la absorbancia y las cantidades relativas de metal y ligando. Si un complejo metal-ligando es muy débil, una gráfica de absorbancia versus X L o n L/n M se vuelve tan curvada que es imposible determinar la estequiometría por extrapolación. En este caso se utiliza la relación pendiente-ratio.
En el método de relación pendiente se preparan dos conjuntos de soluciones. El primer conjunto de soluciones contiene una cantidad constante de metal y una cantidad variable de ligando, elegida de tal manera que la concentración total de metal, C M, es mucho mayor que la concentración total de ligando, C L. Bajo estas condiciones podemos suponer que esencialmente todo el ligando reacciona para formar el complejo metal-ligando. La concentración del complejo, que tiene la forma general M x L y, es
\[\left[\mathrm{M}_{x} \mathrm{L_y}\right]=\frac{C_{\mathrm{L}}}{y} \nonumber\]
Si monitoreamos la absorbancia a una longitud de onda donde solo M x L y absorbe, entonces
\[A=\varepsilon b\left[\mathrm{M}_{x} \mathrm{L}_{y}\right]=\frac{\varepsilon b C_{\mathrm{L}}}{y} \nonumber\]
y una gráfica de absorbancia versus CL es lineal con una pendiente, s L, de
\[s_{\mathrm{L}}=\frac{\varepsilon b}{y} \nonumber\]
Se prepara un segundo conjunto de soluciones con una concentración fija de ligando que es mucho mayor que una concentración variable de metal; así
\[\left[\mathrm{M}_{x} \mathrm{L}_{y}\right]=\frac{C_{\mathrm{M}}}{x} \nonumber\]
\[A=\varepsilon b\left[\mathrm{M}_{x} \mathrm{L}_{y}\right]=\frac{\varepsilon b C_{\mathrm{M}}}{x} \nonumber\]
\[s_{M}=\frac{\varepsilon b}{x} \nonumber\]
Una relación de las pendientes proporciona los valores relativos de x e y.
\[\frac{s_{\text{M}}}{s_{\text{L}}}=\frac{\varepsilon b / x}{\varepsilon b / y}=\frac{y}{x} \nonumber\]
Una suposición importante en el método de relación pendiente es que la reacción de complejación continúa completándose en presencia de un exceso suficientemente grande de metal o ligando. El método de relación pendiente también se limita a sistemas en los que solo se forma un único complejo y para los que se obedece la ley de Beer.
Determinación de Constantes de Equilibrio
Otra aplicación importante de la espectroscopia de absorción molecular es la determinación de constantes de equilibrio. Consideremos, como ejemplo sencillo, una reacción ácido-base de la forma general
\[\operatorname{HIn}(a q)+ \ \mathrm{H}_{2} \mathrm{O}(l) \rightleftharpoons \ \mathrm{H}_{3} \mathrm{O}^{+}(a q)+\operatorname{In}^{-}(a q) \nonumber\]
donde Hin e In son las formas conjugadas de ácido débil y base débil de un indicador ácido-base. La constante de equilibrio para esta reacción es
\[K_{\mathrm{a}}=\frac{\left[\mathrm{H}_{3} \mathrm{O}^{+}\right][\mathrm{A^-}]}{[\mathrm{HA}]} \nonumber\]
Para determinar el valor de la constante de equilibrio, preparamos una solución en la que la reacción se encuentra en estado de equilibrio y determinamos la concentración de equilibrio para H 3 O +, HIn e In —. La concentración de H 3 O + es fácil de determinar midiendo el pH de la solución. Para determinar la concentración de hIN e In, podemos medir la absorbancia de la solución.
Si tanto Hin como In absorben a la longitud de onda seleccionada, entonces, de la ley de Beer, sabemos que
donde\(\varepsilon_\text{HIn}\) y\(\varepsilon_{\text{In}}\) son las absorbilidades molares para Hin e In —. La concentración total del indicador, C, viene dada por una ecuación de balance de masas
\[C=[\mathrm{HIn}]+ [\text{In}^-] \label{10.6}\]
Resolviendo la ecuación\ ref {10.6} para [hIN] y sustituyendo en la ecuación\ ref {10.5} da
\[A=\varepsilon_{\mathrm{Hln}} b\left(C-\left[\mathrm{In}^{-}\right]\right)+\varepsilon_{\mathrm{ln}} b\left[\mathrm{In}^{-}\right] \nonumber\]
que simplificamos
\[A=\varepsilon_{\mathrm{Hln}} bC- \varepsilon_{\mathrm{Hln}}b\left[\mathrm{In}^{-}\right]+\varepsilon_{\mathrm{ln}} b\left[\mathrm{In}^{-}\right] \nonumber\]
donde A hIN, que es igual a\(\varepsilon_\text{HIn}bC\), es la absorbancia cuando el pH es lo suficientemente ácido como para que esencialmente todo el indicador esté presente como hIN. Resolviendo Ecuación\ ref {10.7} para la concentración de In — da
Procediendo de la misma manera, derivamos una ecuación similar para la concentración de hIN
donde A In, que es igual a\(\varepsilon_{\text{In}}bC\), es la absorbancia cuando el pH es lo suficientemente básico como para que solo In — contribuya a la absorbancia. Sustituyendo la ecuación\ ref {10.8} y la ecuación\ ref {10.9} en la expresión constante de equilibrio para hIN da
Podemos usar la Ecuación\ ref {10.10} para determinar K a de una de dos maneras. El enfoque más sencillo es preparar tres soluciones, cada una de las cuales contiene la misma cantidad, C, de indicador. El pH de una solución se hace suficientemente ácido de tal manera que [hIN] >> [In —]. La absorbancia de esta solución da A hIN. El valor de A In se determina ajustando el pH de la segunda solución de tal manera que [In —] >> [hIN]. Finalmente, el pH de la tercera solución se ajusta a un valor intermedio, y se registra el pH y absorbancia, A. El valor de Ka se calcula usando la Ecuación\ ref {10.10}.
La constante de acidez para un indicador ácido-base se determina preparando tres soluciones, cada una de las cuales tiene una concentración total de indicador igual a\(5.00 \times 10^{-5}\) M. La primera solución se hace fuertemente ácida con HCl y tiene una absorbancia de 0.250. La segunda solución se hace fuertemente básica y tiene una absorbancia de 1.40. El pH de la tercera solución es 2.91 y tiene una absorbancia de 0.662. ¿Cuál es el valor de K a para el indicador?
Solución
El valor de Ka se determina haciendo sustituciones apropiadas en 10.20 donde [H 3 O +] es\(1.23 \times 10^{-3}\); así
\[K_{\mathrm{a}}=\left(1.23 \times 10^{-3}\right) \times \frac{0.662-0.250}{1.40-0.662}=6.87 \times 10^{-4} \nonumber\]
Para determinar la K a de un colorante de merocianina, se midió la absorbancia de una solución de colorante\(3.5 \times 10^{-4}\) M a un pH de 2.00, un pH de 6.00 y un pH de 12.00, produciendo absorbancias de 0.000, 0.225 y 0.680, respectivamente. ¿Cuál es el valor de K a para este tinte? Los datos para este problema se adaptan de Lu, H.; Rutan, S. C. Anal. Chem. , 1996, 68, 1381—1386.
- Contestar
-
El valor de K a es
\[K_{\mathrm{a}}=\left(1.00 \times 10^{-6}\right) \times \frac{0.225-0.000}{0.680-0.225}=4.95 \times 10^{-7} \nonumber\]
Un segundo enfoque para determinar Ka es preparar una serie de soluciones, cada una de las cuales contiene la misma cantidad de indicador. Se utilizan dos soluciones para determinar los valores de A hIN y A In. Tomando el log de ambos lados de la Ecuación\ ref {10.10} y reordenando nos dejan con la siguiente ecuación.
Una gráfica de log [(A — A hIN)/(A In — A)] versus pH es una línea recta con una pendiente de +1 y una intersección y de —p K a.
Para determinar la K a para el indicador azul de bromotimol, se mide la absorbancia de cada una serie de soluciones que contienen la misma concentración de azul de bromotimol a niveles de pH de 3.35, 3.65, 3.94, 4.30 y 4.64, produciendo valores de absorbancia de 0.170, 0.287, 0.411, 0.562 y 0.670 , respectivamente. La acidificación de la primera solución a un pH de 2 cambia su absorbancia a 0.006, y ajustando el pH de la última solución a 12 cambia su absorbancia a 0.818. ¿Cuál es el valor de K a para el azul de bromotimol? Los datos para este problema son de Patterson, G. S. J. Chem. Educ. , 1999, 76, 395—398.
- Contestar
-
Para determinar Ka utilizamos la Ecuación\ ref {10.11}, trazando log [(A — A hIN)/(A In — A)] versus pH, como se muestra a continuación.

Ajustar una línea recta a los datos da un modelo de regresión de
\[\log \frac{A-A_{\mathrm{HIn}}}{A_{\mathrm{ln}}-A}=-3.80+0.962 \mathrm{pH} \nonumber\]
La intersección y es —p K a; así, la p K a es 3.80 y la K a es\(1.58 \times 10^{-4}\).
Al desarrollar estos enfoques para determinar K a consideramos un sistema relativamente simple en el que la absorbancia de hIN e In — son fáciles de medir y para el cual es fácil determinar la concentración de H 3 O +. Además de las reacciones ácido-base, podemos adaptar estos enfoques a cualquier reacción de la forma general
\[X(a q)+Y(a q)\rightleftharpoons Z(a q) \nonumber\]
incluyendo reacciones de complejación metal-ligando y reacciones redox, siempre que podamos determinar espectrofotométricamente la concentración del producto, Z, y uno de los reactivos, ya sea X o Y, y que podamos determinar la concentración del otro reactivo por algunos otro método. Con las modificaciones apropiadas, también es posible un sistema más complicado en el que no se pueda determinar la concentración de uno o más de los reactivos o productos [Ramette, R. W. Chemical Equilibrium and Analysis, Addison-Wesley: Reading, MA, 1981, Capítulo 13].
Evaluación de espectroscopía UV/Vis e IR
Escala de Operaciones
La absorción molecular de UV/Vis se utiliza rutinariamente para el análisis de analitos traza en muestras macro y meso. Los analitos mayores y menores se determinan diluyendo la muestra antes del análisis, y la concentración de una muestra puede permitir el análisis de analitos de ultratraza. La escala de operaciones para la absorción infrarroja es generalmente más pobre que la de la absorción UV/Vis.
Precisión
En condiciones normales es fácil obtener un error relativo de 1— 5% con absorción UV/Vis. La precisión generalmente está limitada por la calidad de la pieza en bruto. Ejemplos del tipo de problemas que se encuentran incluyen la presencia de partículas en la muestra que dispersan la radiación, y la presencia de interferentes que reaccionan con reactivos analíticos. En este último caso el interferente puede reaccionar para formar una especie absorbente, lo que conduce a un error determinado positivo. Los interferentes también pueden evitar que el analito reaccione, lo que conduce a un error determinado negativo. Con cuidado, es posible mejorar la precisión de un análisis tanto como un orden de magnitud.
Precisión
En la espectroscopia de absorción, la precisión está limitada por errores indeterminados, principalmente el ruido instrumental, que se introducen cuando medimos la absorbancia. La precisión generalmente es peor para absorbancias bajas donde P 0 ≈ P T, y para absorbancias altas donde P T se acerca a 0. Podríamos esperar, por lo tanto, que la precisión variará con la transmitancia.
Podemos derivar una expresión entre precisión y transmitancia aplicando la propagación de la incertidumbre como se describe en el Capítulo 4. Para ello reescribimos la ley de Beer como
\[C=-\frac{1}{\varepsilon b} \log T \label{10.12}\]
La Tabla 4.3.1 del Capítulo 4 nos ayuda a completar la propagación de la incertidumbre para la Ecuación\ ref {10.12}; así, la incertidumbre absoluta en la concentración, s C, es
\[s_{c}=-\frac{0.4343}{\varepsilon b} \times \frac{s_{T}}{T} \label{10.13}\]
donde s T es la incertidumbre absoluta en la transmitancia. Dividir la ecuación\ ref {10.13} por la ecuación\ ref {10.12} da la incertidumbre relativa en la concentración, s C/C, como
\[\frac{s_c}{C}=\frac{0.4343 s_{T}}{T \log T} \nonumber\]
Si conocemos la incertidumbre absoluta de la transmitancia, entonces podemos determinar la incertidumbre relativa en la concentración para cualquier transmitancia medida.
Determinar la incertidumbre relativa en la concentración es complicado porque s T es una función de la transmitancia. Como se muestra en la Tabla 10.3.3 , se observan tres categorías de error instrumental indeterminado [Rothman, L. D.; Crouch, S. R.; Ingle, J. D. Jr. Anal. Chem. 1975, 47, 1226—1233]. Se observa una constante s T para la incertidumbre asociada a la lectura del% T en una escala analógica o digital de un medidor. Los valores típicos son ±0.2— 0.3% (un k 1 de ±0.002—0.003) para una escala analógica y ± 0.001% a (k 1 de ±0.00001) para una escala digital.
También se observa una constante s T para los transductores térmicos utilizados en espectrofotómetros infrarrojos. El efecto de una constante s T sobre la incertidumbre relativa en la concentración se muestra mediante la curva A en la Figura 10.3.16 . Obsérvese que la incertidumbre relativa es muy grande tanto para absorbancias altas como para absorbancias bajas, alcanzando un mínimo cuando la absorbancia es de 0.4343. Esta fuente de error indeterminado es importante para los espectrofotómetros infrarrojos y para los espectrofotómetros UV/Vis económicos. Para obtener una incertidumbre relativa en la concentración de ±1— 2%, la absorbancia se mantiene dentro del rango 0.1—1.

Los valores de s T son una función compleja de transmitancia cuando los errores indeterminados están dominados por el ruido asociado a los detectores de fotones. La curva B en la Figura 10.3.16 muestra que la incertidumbre relativa en la concentración es muy grande para absorbancias bajas, pero es menor a absorbancias más altas. Aunque la incertidumbre relativa alcanza un mínimo cuando la absorbancia es 0.963, hay poco cambio en la incertidumbre relativa para absorbancias entre 0.5 y 2. Esta fuente de error indeterminado generalmente limita la precisión de los espectrofotómetros UV/Vis de alta calidad para absorbancias medias a altas.
Finalmente, el valor de s T es directamente proporcional a la transmitancia para errores indeterminados que resultan de las fluctuaciones en la intensidad de la fuente y de la incertidumbre en el posicionamiento de la muestra dentro del espectrómetro. Esto último es particularmente importante porque las propiedades ópticas de una celda de muestra no son uniformes. Como resultado, el reposicionamiento de la célula de muestra puede conducir a un cambio en la intensidad de la radiación transmitida. Como muestra la curva C en la Figura 10.3.16 , el efecto es importante solo a bajas absorbancias. Esta fuente de errores indeterminados suele ser el factor limitante para espectrofotómetros UV/Vis de alta calidad cuando la absorbancia es relativamente pequeña.
Cuando la incertidumbre relativa en la concentración está limitada por la resolución de lectura del% T, es posible mejorar la precisión del análisis redefiniendo 100% T y 0% T. Normalmente se establece 100% T usando un blanco y 0% T se establece mientras se evita que la radiación de la fuente alcance el detector. Si la absorbancia es demasiado alta, se mejora la precisión restableciendo 100% T usando una solución estándar de analito cuya concentración es menor que la de la muestra (Figura 10.3.17 a). Para una muestra cuya absorbancia es demasiado baja, se mejora la precisión al redefinir 0% T usando una solución estándar del analito cuya concentración es mayor que la del analito (Figura 10.3.17 b). En este caso se requiere una curva de calibración debido a que ya no existe una relación lineal entre absorbancia y concentración. La precisión se incrementa aún más al combinar estos dos métodos (Figura 10.3.17 c). Nuevamente, es necesaria una curva de calibración ya que la relación entre absorbancia y concentración ya no es lineal.

Sensibilidad
La sensibilidad de un método de absorción molecular, que es la pendiente de una curva de calibración de la ley de Beer, es el producto de la absortividad del analito y la longitud de trayectoria de la célula de muestra (\(\varepsilon b\)). Puede mejorar la sensibilidad de un método seleccionando una longitud de onda donde la absorbancia esté en un máximo o aumentando la longitud de la ruta.
Consulte la Figura 10.2.10 para ver un ejemplo de cómo la elección de la longitud de onda afecta la sensibilidad de una curva de calibración.
Selectividad
La selectividad rara vez es un problema en la espectrofotometría de absorción molecular. En muchos casos es posible encontrar una longitud de onda donde solo el analito absorba. Cuando dos o más especies contribuyen a la absorbancia medida, todavía es posible un análisis multicomponente, como se muestra en Ejemplo 10.3.2 y Ejemplo 10.3.3 .
Tiempo, Costo y Equipo
El análisis de una muestra por espectroscopía de absorción molecular es relativamente rápido, aunque se requiere tiempo adicional si necesitamos convertir un analito no absorbente en una forma absorbente. El costo de la instrumentación UV/Vis varía de varios cientos de dólares para un fotómetro de filtro simple, a más de $50,000 para un instrumento de doble haz de alta resolución controlado por computadora equipado con anchos de ranura variables y que opera en un rango extendido de longitudes de onda. Los espectrómetros infrarrojos con transformada de Fourier se pueden obtener por tan solo $15,000—$20,000, aunque hay modelos más caros disponibles.