
11.13: El cero absoluto es inalcanzable
- Page ID
- 73862

La tercera ley postula que la entropía de una sustancia es siempre finita y que se acerca a una constante a medida que la temperatura se acerca a cero. El valor de esta constante es independiente de los valores de cualquier otra función estatal que caracterice a la sustancia. Para cualquier sustancia dada, somos libres de asignar un valor seleccionado arbitrariamente al valor límite de temperatura cero. Sin embargo, no podemos asignar entropías arbitrarias de temperatura cero a todas las sustancias. El conjunto de asignaciones que hagamos debe ser consistente con los valores limitantes de temperatura cero observados experimentalmente de los cambios de entropía de las reacciones entre diferentes sustancias. Para sustancias perfectamente cristalinas, estas entropías de reacción son todas cero. Podemos satisfacer esta condición asignando un valor arbitrario a la entropía molar de temperatura cero de cada elemento y estipulando que la entropía de temperatura cero de cualquier compuesto es la suma de las entropías de temperatura cero de sus elementos constituyentes. Este cálculo se simplifica enormemente si dejamos que la entropía de temperatura cero de cada elemento sea cero. Este es el contenido esencial de la tercera ley.
La declaración Lewis y Randall incorpora esta selección del estado de referencia de entropía cero para las entropías, especificándolo como “un estado cristalino” de cada elemento a cero grados. Como resultado, la entropía de cualquier sustancia a cero grados es mayor o igual a cero. Es decir, la declaración de Lewis y Randall incluye una convención que fija el valor límite de temperatura cero de la entropía de cualquier sustancia. Al respecto, la declaración de Lewis y Randall hace una elección esencialmente arbitraria que no es una propiedad intrínseca de la naturaleza. Vemos, sin embargo, que es una elección abrumadoramente conveniente.
Hemos discutido declaraciones alternativas de las leyes primera y segunda. También son posibles varias declaraciones alternativas de la tercera ley. Consideramos lo siguiente:
Es imposible lograr una temperatura de cero absoluto.
Esta declaración es más general que la declaración de Lewis y Randall. Si consideramos la aplicación de esta afirmación a las temperaturas alcanzables en procesos que involucran una sola sustancia, podemos demostrar que implica, y está implícito por, la afirmación de Lewis y Randall.
Las propiedades de la capacidad calorífica\(C_P\),, juegan un papel central en estos argumentos. Hemos visto que\(C_P\) es una función de la temperatura. Si bien no es útil hacerlo, podemos aplicar la relación definitoria para\(C_P\) a una sustancia que experimenta una transición de fase y encontrar\(C_P=\infty\). Si pensamos en una sustancia cuya capacidad calorífica es inferior a cero, nos encontramos con una contradicción de nuestras ideas básicas sobre el calor y la temperatura: Si\(q>0\) y\({q}/{\Delta T}<0\), debemos tener\(\Delta T<0\); es decir, calentar la sustancia hace que su temperatura disminuya. En definitiva, la teoría que hemos desarrollado incrusta premisas que requieren\(C_P>0\) para cualquier sistema sobre el que podamos realizar mediciones.
Caracterizemos un sistema de sustancias puras por su presión y temperatura y consideremos procesos reversibles de presión constante en los que solo es posible trabajar a presión y volumen. Entonces\({\left({\partial S}/{\partial T}\right)}_P={C_P}/{T}\) y\(dS={C_PdT}/{T}\). Ahora queremos mostrar: la estipulación de Lewis y Randall de que la entropía es siempre finita requiere que la capacidad calorífica vaya a cero cuando la temperatura vaya a cero. (Ya que vamos a demostrar que la tercera ley prohíbe las mediciones a cero absoluto, esta conclusión es congruente con nuestra conclusión del párrafo anterior.) Que la capacidad calorífica va a cero cuando la temperatura va a cero es evidente a partir de\(S={C_PdT}/{T}.\) Si\(C_P\) no va a cero cuando la temperatura va a cero,\(dS\) se vuelve arbitrariamente grande ya que la temperatura va a cero, lo que contradice la afirmación de Lewis y Randall.
Para desarrollar este resultado de manera más explícita, dejamos que las capacidades de calor a temperaturas\(T\) y cero sean\(C_P\left(T\right)\) y\(C_P\left(0\right)\), respectivamente. Ya que\(C_P\left(T\right)>0\) para cualquiera\(T\ >\ 0\), tenemos\(S\left(T\right)-S\left(T^*\right)>0\) para cualquiera\(T>T^*>0\). Ya que la entropía es siempre finita,\(\infty >S\left(T\right)-S\left(T^*\right)>0\), así que
\[\infty >{\mathop{\mathrm{lim}}_{T^*\to 0} \left[S\left(T\right)-S\left(T^*\right)\right]\ }>0\]
y
\[\infty >{\mathop{\mathrm{lim}}_{T^*\to 0} \int^T_{T^*}{\frac{C_P}{T}}\ }dT>0\]
Para temperaturas cercanas a cero, podemos expandir la capacidad calorífica, con precisión arbitraria, como polinomio de la serie Taylor en\(T\):
\[C_P\left(T\right)=C_P\left(0\right)+\left(\frac{\partial C_P\left(0\right)}{\partial T}\right)_PT +\frac{1}{2} \left(\frac{{\partial }^2C_P\left(0\right)}{\partial T^2}\right)_PT^2+\dots \]
Las desigualdades se convierten
\[\infty >{\mathop{\mathrm{lim}}_{T^*\to 0} \left\{C_P\left(0\right){ \ln \frac{T}{T^*}\ }+{\left(\frac{\partial C_P\left(0\right)}{\partial T}\right)}_P\left(T-T^*\right)+\frac{1}{4}{\left(\frac{{\partial }^2C_P\left(0\right)}{\partial T^2}\right)}_P{\left(T-T^*\right)}^2+\dots \right\}\ }>0\]
El estado de la izquierda requiere\(C_P\left(0\right)=0\).
Podríamos ver la tercera ley como una declaración sobre las capacidades térmicas de las sustancias puras. Inferimos no solo eso\(C_P>0\) para todos\(T>0\), sino también que
\[{\mathop{\mathrm{lim}}_{T\to 0} \left(\frac{C_P}{T}\right)=0\ }\]
De manera más general, podemos inferir aseveraciones correspondientes para sistemas reversibles cerrados que no son sustancias puras:\({\left({\partial H}/{\partial T}\right)}_P>0\) para todos\(T>0\), y\({\mathop{\mathrm{lim}}_{T\to 0} T^{-1}{\left({\partial H}/{\partial T}\right)}_P=0\ }\). (Sin embargo, las entropías de temperatura cero de tales sistemas no son cero). En la discusión a continuación, describimos el sistema como una sustancia pura. Podemos hacer esencialmente los mismos argumentos para cualquier sistema; sólo necesitamos reemplazarlos\(C_P\) por\({\left({\partial H}/{\partial T}\right)}_P\). El enunciado de Lewis y Randall afirma que la entropía va a una constante en cero absoluto, independientemente de los valores de cualquier otra función termodinámica. De ello se deduce que la entropía a cero grados es independiente del valor de la presión. Para dos presiones cualesquiera,\(P_1\) y\(P_2\), tenemos\(S\left(P_2,0\right)-S\left(P_1,0\right)=0\). Dejando\({P=P}_1\)\(P_2=P+\Delta P\) y y, tenemos
\[\frac{S\left(P+\Delta P,0\right)-S\left(P,0\right)}{\Delta P}=0\]
para cualquier\(\Delta P\). Por lo tanto, tenemos
\[{\left(\frac{\partial S}{\partial P}\right)}_{T=0}=0\]
En el Capítulo 10, encontramos\({\left({\partial S}/{\partial }P\right)}_T=-{\left({\partial V}/{\partial T}\right)}_P\), por lo que tanto la entropía como el volumen se acercan asintóticamente a sus valores de temperatura cero.
Cuando decimos que el cero absoluto es inalcanzable, queremos decir que ningún sistema puede sufrir ningún cambio en el que su temperatura final sea cero. Para ver por qué el cero absoluto debe ser inalcanzable, consideremos procesos que puedan disminuir la temperatura de un sistema. En general, tenemos depósitos de calor disponibles a diversas temperaturas. Podemos seleccionar el reservorio disponible cuya temperatura es más baja, y llevar el sistema a esta temperatura por simple contacto térmico. Esto es trivial; claramente, el reto es disminuir aún más la temperatura. Para ello, debemos efectuar algún otro cambio. Sea cual sea este cambio, no puede ser ayudado por un intercambio de calor con el entorno. Una vez que hemos llevado el sistema a la temperatura de la porción más fría disponible del entorno, cualquier intercambio adicional de calor con el entorno solo puede ser contraproducente. Concluimos que cualquier proceso adecuado a nuestro propósito debe ser adiabático. Dado que un proceso adiabático no intercambia calor con el entorno,\(\Delta \hat{S}=0\).
El proceso también debe ser un proceso posible, de manera que\(\Delta S+\Delta \hat{S}\ge 0\), y como es adiabático,\(\Delta S\ge 0\). Consideremos un proceso reversible y un proceso irreversible en el que el mismo sistema\({}^{2}\) pasa del estado especificado por\(P_1\) y\(T_1\) a un segundo estado en el que se encuentra la presión\(P_2\). Las temperaturas finales y los cambios de entropía de estos procesos son diferentes. Para el proceso reversible,\(\Delta S=0\); designamos la temperatura final como\(T_2\). Para el proceso irreversible,\(\Delta S>0\); designamos la temperatura final como\(T^*_2\). Resulta que el cambio de temperatura es menor para el proceso irreversible que para el proceso reversible; es decir,\(T_2-T_1<t^*_2-t_1\) >. Equivalentemente, el proceso reversible alcanza una temperatura más baja:\({T_2<t}^*_2\) >. Desde
\[dS=\frac{C_P}{T}dT-{\left(\frac{\partial V}{\partial T}\right)}_PdP\]
podemos calcular los cambios de entropía para estos procesos. Para el proceso reversible, calculamos\[\Delta S^{rev}=S\left(P_2,T_2\right)-S\left(P_1,T_1\right)\]
Para ello, primero calculamos
\[{\left(\Delta S\right)}_T=S\left(P_2,T_1\right)-S\left(P_1,T_1\right)\]
para la transformación reversible isotérmica de estado\(P_1\),\(T_1\) al estado especificado por\(P_2\) y\(T_1\). Para este paso,\(dT\) es cero, y así
\[{\left(\Delta S\right)}_T=\int^{P_2}_{P_1}{{\left(\frac{\partial V}{\partial T}\right)}_PdP}\]
Luego calculamos
\[{\left(\Delta S\right)}_P=S\left(P_2,T_2\right)-S\left(P_2,T_1\right)\]
para la transformación reversible isobárica de estado\(P_2\),\(T_1\) a estado\(P_2\),\(T_2\). Para esta transformación,\(dP\) es cero, y
\[{\left(\Delta S\right)}_P=-\int^{T_2}_{T_1}{\frac{C_P}{T}dT}\]
Entonces,
\[\Delta S^{rev}=S\left(P_2,T_2\right)-S\left(P_1,T_1\right)=\int^{T_2}_{T_1}{\frac{C_P}{T}dT}-\int^{P_2}_{P_1}{{\left(\frac{\partial V}{\partial T}\right)}_PdP}=0\]
Porque\(\Delta S^{rev}=0\), el proceso reversible es único; es decir\(P_1\), dado\(T_1\), y\(P_2\), se determina la temperatura final del sistema. Nos encontramos\(T_2\) desde
\[\int^{T_2}_{T_1}{\frac{C_P}{T}dT}=\int^{P_2}_{P_1}{{\left(\frac{\partial V}{\partial T}\right)}_PdP}\]
Para entender el cambio de entropía para el proceso irreversible, observamos primero que hay un número infinito de tales procesos. No hay nada único en la temperatura final. Dada\(P_1\),\(T_1\), y\(P_2\), la temperatura final,\(T^*_2\), puede tener cualquier valor consistente con las propiedades de la sustancia. Para especificar un proceso irreversible particular, debemos especificar las cuatro cantidades\(P_1\),\(T_1\)\(P_2\), y\(T^*_2\). Habiéndolo hecho, sin embargo, podemos calcular el cambio de entropía para el proceso irreversible,
\[\Delta S^{irrev}=S\left(P_2,T^*_2\right)-S\left(P_1,T_1\right)>0\]
calculando los cambios de entropía a medida que transportamos reversiblemente el sistema a lo largo del camino isotérmico de dos pasos desde\(P_1\),\(T_1\) hasta\(P_2\),\(T_1\) y luego a lo largo de la ruta isobárica desde\(P_2\),\(T_1\) hasta\(P_2\),\(T^*_2\). El cálculo de\(\Delta S^{irrev}\) para esta ruta reversible de\(P_1\),\(T_1\) a\(P_2\),\(T^*_2\) emplea la misma lógica que el cálculo, en el párrafo anterior, de\(\Delta S\) para el camino reversible de\(P_1\),\(T_1\) a\(P_2\),\(T_2\). La diferencia es que\(T^*_2\) reemplaza\(T_2\) como límite superior en la integral de temperatura. La integral de presión es la misma. Tenemos
\[\Delta S^{irrev}=S\left(P_2,T^*_2\right)-S\left(P_1,T_1\right)=\int^{T^*_2}_{T_1}{\frac{C_P}{T}dT}-\int^{P_2}_{P_1}{{\left(\frac{\partial V}{\partial T}\right)}_PdP} >0\]
Desde\(\Delta S^{irrev}>\Delta S^{rev}\), tenemos
\[\int^{T^*_2}_{T_1}{\frac{C_P}{T}dT}>\int^{T_2}_{T_1}{\frac{C_P}{T}dT}\]
Dado que los integrandos son los mismos y positivos, de ello se deduce que\(T^*_2>T_2\), como se aseveró anteriormente.
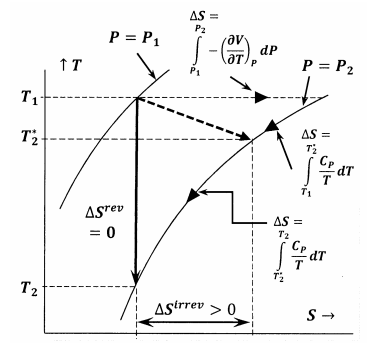
En la Figura 6 se muestran las relaciones entre las diversas cantidades discutidas en este argumento. En la primera instancia, la Figura 6 muestra una gráfica de dos de las isobarras del sistema en el espacio de temperatura-entropía. Es decir, la línea etiquetada\(P=P_1\) representa el conjunto de puntos de temperatura-entropía en los que el sistema equilibrado tiene presión\(P_1\); la línea etiquetada\(P=P_2\) representa las posiciones de equilibrio a presión\(P_2\). Otras líneas en este boceto representan caminos a lo largo de los cuales el sistema puede sufrir cambios reversibles a entropía constante o temperatura constante. La línea punteada representa el proceso irreversible en el que el sistema va del estado especificado por\(P_1\),\(T_1\) al estado especificado por\(P_2\),\(T^*_2\). Esta línea está punteada para representar el hecho de que la temperatura del sistema puede no estar bien definida durante el proceso irreversible.
El enfriamiento efectivo se puede lograr usando cambios de presión si el sistema es un gas. Sin embargo, para líquidos y sólidos,\({\left({\partial V}/{\partial T}\right)}_P\) es pequeño; en consecuencia, el cambio de temperatura para un cambio de presión reversible también es pequeño. A temperaturas cercanas al cero absoluto, casi todas las sustancias son sólidas; para lograr un enfriamiento efectivo debemos cambiar una variable termodinámica para la cual el coeficiente de temperatura de un sólido es lo más grande posible. Para considerar el problema general de disminuir la temperatura de un sistema variando algo que no sea la presión, debemos considerar un sistema en el que sea posible alguna forma de trabajo sin presión-volumen. Tal sistema está sujeto a una fuerza adicional, y su energía cambia a medida que cambia esta fuerza.
Desmagnetización adiabática
El método práctico mediante el cual se logran temperaturas extremadamente bajas se denomina desmagnetización adiabática. Este método explota las propiedades de los sólidos paramagnéticos. En tales sólidos, los electrones desapareados localizados en átomos individuales dan lugar a un momento magnético. La mecánica cuántica lleva a conclusiones importantes sobre la interacción entre dichos momentos magnéticos y un campo magnético aplicado: En un campo magnético aplicado, se cuantifica el momento magnético de un átomo individual. En el caso más simple, puede alinearse en solo dos direcciones; debe ser paralela o antiparalela al campo magnético aplicado. Cuando el momento magnético de un átomo es paralelo al campo magnético, la energía del sistema es menor que cuando la alineación es antiparalela. El campo magnético aplicado ejerce una fuerza sobre los momentos magnéticos asociados con átomos individuales. La energía del sistema depende de la magnitud del campo magnético aplicado.
En lugar de enfocarnos en el caso particular de la desmagnetización adiabática, consideremos los cambios de energía y entropía asociados con cambios en un potencial generalizado\({\mathit{\Phi}}_{\theta }\), y su desplazamiento generalizado,\(\theta\). (Para la desmagnetización adiabática,\(\theta\) sería el campo magnético aplicado.) Se requieren tres variables para describir los cambios reversibles en este sistema. Podemos expresar la energía y la entropía como funciones de temperatura, presión y\(\theta\):
\(E=E\left(T,P,\theta \right)\)y\(S=S\left(T,P,\theta \right)\). El diferencial total de la entropía incluye un término que especifica la dependencia de la entropía de\(\theta\). Tenemos
\[dS={\left(\frac{\partial S}{\partial T}\right)}_{P,\theta }dT+{\left(\frac{\partial S}{\partial P}\right)}_{T,\theta }dP+{\left(\frac{\partial S}{\partial \theta }\right)}_{T,P}d\theta =\frac{C\left(T,P,\theta \right)}{T}dT-{\left(\frac{\partial V}{\partial T}\right)}_{P,\theta }dP+{\left(\frac{\partial S}{\partial \theta }\right)}_{T,P}d\theta\]
donde escribimos\(C\left(T,P,\theta \right)\) para enfatizar que nuestros propósitos actuales requieren ahora que medimos la capacidad calorífica a presión constante y constante\(\theta\).
Para presión constante, P, y desplazamiento constante,\(\theta\), la entropía depende de la temperatura como
\[S\left(T,P,\theta \right)=S\left(0,P,\theta \right)+\int^T_0 \left(\frac{\partial S}{\partial T}\right)_{P,\theta }dT=S\left(0,P,\theta \right)+\int^T_0 \frac{C\left(T,P,\theta \right)}{T}dT\]
El postulado de que la entropía sea finita a cualquier temperatura implica que la capacidad calorífica\(\theta\) dependiente de presión y presión se convierte en cero a cero absoluto. Es decir, a cero absoluto, la capacidad calorífica se desvanece cualesquiera que sean los valores de P y\(\theta\). El argumento es exactamente el mismo que antes. Antes, escribimos\(C_P\left(0\right)=0\); para el presente caso generalizado, escribimos\(C\left(0,P,\theta \right)=0\).
De igual manera, del postulado de que la entropía va a una constante a cero absoluto para todos los valores de las demás variables termodinámicas, se deduce que, para dos presiones cualesquiera\(P_1\) y\(P_2\), y para dos valores cualesquiera del desplazamiento generalizado,\({\theta }_1\) y\({\theta }_2\),
\[S\left(0,P_1,{\theta }_1\right)=S\left(0,P_2,{\theta }_1\right)=S\left(0,P_1,{\theta }_2\right)=S\left(0,P_2,{\theta }_2\right)=0\]
y de ahí que
\[{\left(\frac{\partial S}{\partial P}\right)}_{T=0,\theta }={\left(\frac{\partial S\left(0,P,\theta \right)}{\partial P}\right)}_{T,\theta }=0\]y\[{\left(\frac{\partial S}{\partial \theta }\right)}_{T=0,P}={\left(\frac{\partial S\left(0,P,\theta \right)}{\partial \theta }\right)}_{T,P}=0\]
Queremos considerar un proceso en el que un sistema pase de la temperatura más baja disponible en los alrededores a una temperatura aún más baja. Para minimizar la temperatura final, este proceso debe realizarse adiabáticamente. También debe ser un proceso posible, así que eso\(dS\ge 0\). Por simplicidad, supongamos ahora que llevamos a cabo este proceso a una presión constante,\(P\), y que el sistema va del estado especificado por\(P\),\(T_1\),\({\theta }_1\) al estado especificado por\(P\),\(T_2\),\({\theta }_2\) donde\(T_1>T_2\). Las entrópías de estos dos estados son
\[S\left(T_1,P,{\theta }_1\right)=S\left(0,P,{\theta }_1\right)+\int^{T_1}_0{\frac{C\left(T,P,{\theta }_1\right)}{T}}dT\]y\[S\left(T_2,P,{\theta }_2\right)=S\left(0,P,{\theta }_2\right)+\int^{T_2}_0{\frac{C\left(T,P,{\theta }_2\right)}{T}}dT\]
El cambio de entropía para este proceso es
\[S\left(T_2,P,{\theta }_2\right)-S\left(T_1,P,{\theta }_1\right)=S\left(0,P,{\theta }_2\right)-S\left(0,P,{\theta }_1\right)\]\[+\int^{T_2}_0{\frac{C\left(T,P,{\theta }_2\right)}{T}}dT-\int^{T_1}_0{\frac{C\left(T,P,{\theta }_1\right)}{T}}dT\ge 0\]
Ahora, supongamos que la temperatura final es cero; es decir,\(T_2=0\), para que
\[\int^{T_2}_0{\frac{C\left(T,P,{\theta }_2\right)}{T}}dT=0\]De ello se deduce que\[S\left(0,P,{\theta }_2\right)-S\left(0,P,{\theta }_1\right)\ge \int^{T_1}_0{\frac{C\left(T,P,{\theta }_1\right)}{T}}dT>0\]
donde la desigualdad a la derecha se deriva del hecho de que eso\(C\left(T,P,{\theta }_1\right)>0\). Entonces, se deduce que
\[S\left(0,P,{\theta }_2\right)-S\left(0,P,{\theta }_1\right)>0\]
que contradice la declaración de Lewis y Randall de la tercera ley. El supuesto de que el sistema puede alcanzar el cero absoluto lleva a una contradicción de la declaración de Lewis y Randall de la tercera ley. Por lo tanto, si la afirmación de Lewis y Randall es verdadera, el cero absoluto es inalcanzable.
También se aplica lo contrario; es decir, a partir de la proposición de que el cero absoluto es inalcanzable, podemos demostrar que la afirmación de Lewis y Randall es cierta. Para ello, reorganizamos la ecuación anterior para\(\Delta S\),
\[\int^{T_2}_0{\frac{C\left(T,P,{\theta }_2\right)}{T}}dT\ge\]\[\int^{T_1}_0{\frac{C\left(T,P,{\theta }_1\right)}{T}}dT-S\left(0,P,{\theta }_2\right)+S\left(0,P,{\theta }_1\right)\]
Si ahora asumimos que la declaración de Lewis y Randall es falsa, la expresión de la derecha puede ser menor o igual a cero. La integral de la izquierda puede ser entonces cero, en cuyo caso el sistema puede alcanzar el cero absoluto. Si la declaración de Lewis y Randall es falsa, es cierto que el sistema puede alcanzar el cero absoluto. Por lo tanto: Si el sistema no puede alcanzar el cero absoluto, la afirmación de Lewis y Randall es cierta.
Las figuras 7 y 8 representan estas ideas usando gráficas de contorno en el espacio de temperatura-entropía. Cada figura muestra dos curvas de nivel. Una de estas líneas de contorno es un conjunto de valores de temperatura y entropía a lo largo de los cuales la presión es constante en\(P\) y\(\theta\) es constante a\({\theta }_1\). La otra línea de contorno es un conjunto de valores de temperatura y entropía a lo largo de los cuales la presión es constante en\(P\) y\(\theta\) es constante a\({\theta }_2\). La pendiente de una línea de contorno es
\[{\left(\frac{\partial T}{\partial S}\right)}_{P,\theta }=\frac{T}{C\left(T,P,\theta \right)}\]
Debido a que la capacidad calorífica siempre es positiva, esta pendiente siempre es positiva.
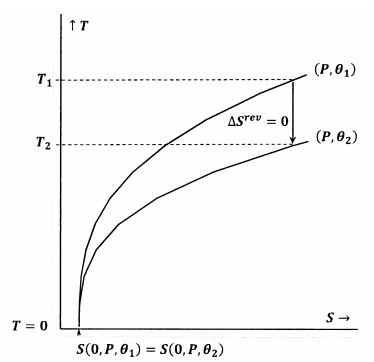
En la Figura 7, queda satisfecha la declaración de Lewis y Randall. Cuando la temperatura llega a cero, las curvas de nivel se encuentran al mismo valor de la entropía; estos contornos satisfacen la relación
\[S\left(0,P,{\theta }_1\right)=S\left(0,P,{\theta }_2\right)\]
Una trayectoria adiabática (vertical) desde el contorno para\(P\) y\({\theta }_1\) cumple con el contorno para\(P\) y\({\theta }_2\) a una temperatura positiva,\(T_2>0\). Dado que esto es evidentemente cierto para cualquiera\(P\) y cualquiera\({\theta }_2\), el estado final para cualquier proceso adiabático tendrá\(T_2>0\). Debido a que la declaración de Lewis y Randall está satisfecha, el sistema no puede alcanzar el cero absoluto, y viceversa.
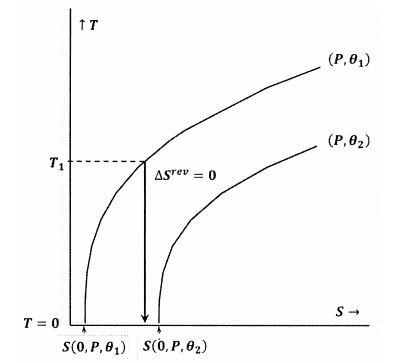
En la Figura 8 se viola la declaración de Lewis y Randall, porque la tenemos\(S\left(0,P,{\theta }_1\right)\). En este caso, un proceso adiabático iniciado a partir de una temperatura inicial suficientemente baja,\(T_1\), alcanzará el cero absoluto sin intersectar el contorno para constantes\(P\) y\({\theta }_2\). Debido a que se viola la declaración de Lewis y Randal, el sistema puede llegar al cero absoluto, y viceversa.