
6.10: Orbitales Moleculares
- Page ID
- 71032

Antes de pasar a discutir métodos que van más allá del modelo de HF, es apropiado examinar parte del esfuerzo computacional que implica llevar a cabo un cálculo de HF SCF en una molécula. Las principales diferencias que aparecen cuando se consideran moléculas en lugar de átomos son
- El Hamiltoniano electrónico\(H_e\) contiene no sólo un potencial Coulomb de atracción nuclear\(\sum_j Ze^2/r_j\), sino una suma de tales términos, uno por cada núcleo de la molécula:
\[\sum_a \sum_j \dfrac{Z_a e^2}{|r_j-R_a|}, \label{6.1.41}\]
cuyas ubicaciones se denotan\(R_a\).
- Uno tiene funciones de base AO del tipo discutido anteriormente localizadas en cada núcleo de la molécula. Estas funciones aún se denotan\(\chi_\mu(r-R_a)\), pero sus dependencias radiales y angulares implican la distancia y orientación del electrón en relación con el núcleo particular en el que se encuentra el AO.
Aparte de estos dos cambios, realizar un cálculo de SCF en una molécula (o ion molecular) procede tal como en el caso atómico detallado anteriormente. Revisemos brevemente cómo se produce este proceso iterativo.
Una vez que se han elegido los conjuntos de bases atómicas para cada átomo, se deben evaluar las integrales de uno\(H_e\) y dos electrones que aparecen en las matrices y superpuestas. Existen numerosos códigos informáticos altamente eficientes que permiten que tales integrales se computen para\(s\)\(p\),\(d\),\(f\), e incluso\(g\)\(h\), y\(i\) funciones básicas. Después de ejecutar uno de estos llamados paquetes integrales para una base con un total de\(M\) funciones, se tiene disponible (generalmente en el disco duro de la computadora) del orden de integrales de\(\dfrac{M^2}{2}\) un electrón (\(\langle \chi_\mu | H_e | \chi_\nu \rangle\)y\(\langle \chi_\mu | \chi_\nu \rangle\)) y\(\dfrac{M^4}{8}\) dos electrones (\(\langle \chi_\mu \chi_\delta | \chi_\nu \chi_\kappa \rangle\)). Al tratar conjuntos de bases orbitales atómicas extremadamente grandes (por ejemplo, 500 o más funciones básicas), los programas informáticos modernos calculan las integrales necesarias, pero nunca las almacenan en el disco. En cambio, sus contribuciones a los elementos de la\(\langle\chi_\mu |H_e|\chi_\nu\rangle\) matriz se acumulan sobre la marcha después de lo cual se descartan las integrales. Esto generalmente se conoce como el enfoque directo integrado.
Formas, Tamaños y Energías de Orbitales
Cada espín-orbital molecular (MO) que resulta de resolver las ecuaciones de HF SCF para una molécula o ion molecular consiste en una suma de componentes que involucran todos los AO básicos:
\[\phi_j = \sum_\mu C_{J,\mu} \chi_\mu.\label{6.1.42}\]
En esta expresión, los\(C_{j,\mu}\) son referidos como coeficientes LCAO-MO porque nos dicen cómo combinar linealmente los AO para formar los MOs. Debido a que los AO tienen varias formas angulares (por ejemplo\(s\)\(p\),,, o\(d\) formas) y extensiones radiales (es decir, diferentes exponentes orbitales), los MO construidos a partir de ellos pueden ser de diferentes formas y tamaños radiales. Veamos algunos ejemplos para ver a qué me refiero.
El primer ejemplo es bastante simple y se refiere a dos átomos de H que se combinan para formar la\(H_2\) molécula. Los AO de valencia en cada átomo de H son los\(1s\) AO; se combinan para formar los dos MO de valencia (\(\sigma\)y\(\sigma^*\)) representados en la Figura 6.1.4.
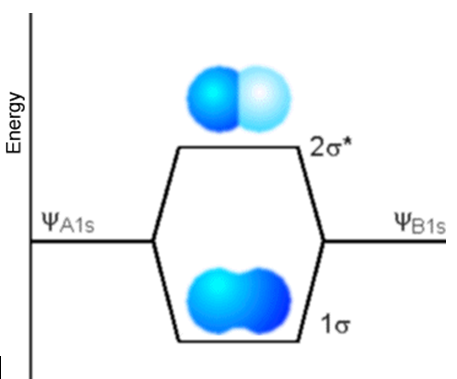
El MO de unión etiquetado s tiene coeficientes LCAO-MO de signo igual para los\(1s\) dos AO, como resultado de lo cual este MO tiene el mismo signo cerca del núcleo H izquierdo (A) que cerca del núcleo H derecho (B). En contraste, el MO antiadhesión etiquetado\(\sigma^*\) tiene coeficientes LCAO-MO de signo diferente para los AO A y\(1s\) B. Como fue el caso en el modelo Hückel o de unión estrecha esbozado en el Capítulo 2, la división de energía entre los dos MO depende del solapamiento\(\langle \chi_{1sA}|\chi_{1sB} \rangle\) entre los dos AOs que, a su vez, depende de la distancia\(R\) entre los dos núcleos.
Un par análogo de MO de unión y antiunión surge cuando dos\(p\) orbitales se superponen lateralmente como en etileno para formar\(\pi\) y\(\pi^*\) MO que se ilustran en la Figura 6.1.5.
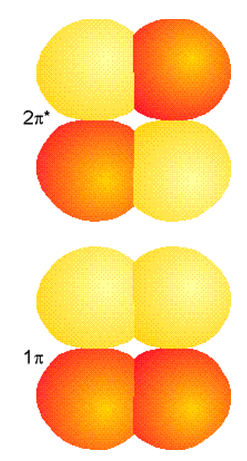
Las formas de estos MO claramente están dictadas por las formas de los AO que los comprenden y los signos relativos de los coeficientes LCAO-MO que relacionan los MO con los AO. Para el\(\pi\) MO, estos coeficientes tienen el mismo signo en los átomos izquierdo y derecho; para el\(\pi^*\) MO, tienen signos opuestos.
Debo enfatizar que los signos y magnitudes de los coeficientes LCAO-MO surgen como vectores propios de la ecuación de valores propios de la matriz HF SCF:
\[\sum_\mu \langle \chi_\nu|h_e| \chi_\mu \rangle C_{j,m} = \epsilon_j \sum_\mu \langle \chi_\nu|\chi_\mu \rangle C_{j,m} \]
Es una característica de tales problemas de autovalor que las funciones propias de menor energía tengan menos nodos que las soluciones de mayor energía como aprendimos de varios ejemplos que resolvimos en la Parte 1 de este texto.
Otra cosa a tener en cuenta sobre los MO mostrados anteriormente es que diferirán en sus detalles cuantitativos, pero no en sus formas generales, cuando varios grupos funcionales se unen a los átomos de C de la molécula de etileno. Por ejemplo, si grupos aceptores de electrones como Cl, OH o Br están unidos a uno de los átomos de C, el potencial atractivo experimentado por un\(\pi\) electrón cerca de ese átomo de C se potenciará en relación con el potencial cercano al otro átomo de C. Como resultado, el MO de unión tendrá mayores coeficientes LCAO-MO Ck, m pertenecientes a AOs de base más apretada\(\chi_\mu\) sobre este átomo de C. Esto hará que el\(\pi\) MO de unión sea más compacto radialmente en esta región del espacio, aunque su carácter nodal y su forma bruta no cambiarán. Alternativamente, un grupo donador de electrones como H 3 C- o t-butilo unido a uno de los centros C hará que el\(\pi\) MO sea más difuso (haciendo que sus coeficientes LCAO-MO para AOs de base más difusa sean más grandes).
Además de los MO formados principalmente por AOs de un tipo (es decir,\(H_2\) porque son principalmente orbitales de tipo s los que forman los\(\sigma\) y\(\sigma^*\) MOs; para el\(\pi\) enlace de etileno, son principalmente los\(2p\) AOs C los que contribuyen), hay MOs de unión y antiunión formados al combinar varios AOs. Por ejemplo, los cuatro MOs equivalentes de unión C-H que\(CH_4\) se muestran en la Figura 6.1. 6 involucran cada uno de los AO con\(1s\) base C\(2s\) y así\(2p\) como H.
Las energías de los MO dependen de dos factores primarios: las energías de los AO a partir de los cuales se construyen los MO y el solapamiento entre estos AO. En la Figura 6.1 se muestra el patrón de energías para los MO de valencia formados por la combinación de pares de átomos de primera fila para formar moléculas diatómicas homo-nucleares.
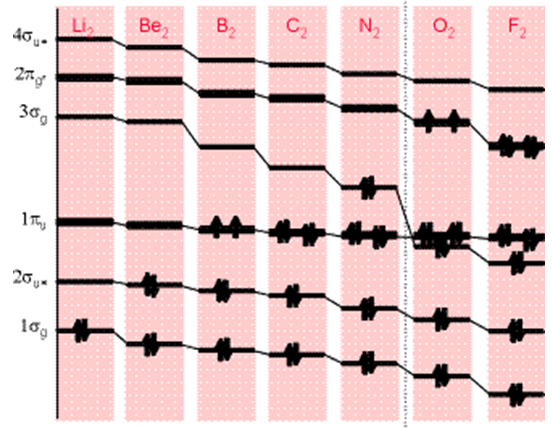
En esta figura, no se muestran los MO centrales formados a partir de\(1s\) los AO; solo aparecen los MO formados\(2s\) y los\(2p\) AO formados a partir de los AO. La clara tendencia hacia energías orbitales más bajas a medida que uno se mueve de izquierda a derecha se debe principalmente a las tendencias en las energías orbitales de los AO constituyentes. Es decir, F siendo más electronegativo que\(N\) tiene un\(2p\) orbital de menor energía que lo hace\(N\).
Orbitales de unión, antiadherencia, no unión y Rydberg
Como se señaló anteriormente, cuando los AO de valencia se combinan para formar MO, los signos relativos de los coeficientes de combinación determinan, junto con las magnitudes de solapamiento AO, la energía del MO y las propiedades nodales. Además de los MO de unión y antiadhesión discutidos e ilustrados anteriormente, es importante conocer otros dos tipos de MO.
Los MO no unidos surgen, por ejemplo, cuando un orbital en un átomo no se dirige hacia y se superpone con un orbital en un átomo vecino. Por ejemplo, los orbitales de pares solitarios sobre\(H_2O\) o sobre el átomo de oxígeno de\(H_2C=O\) son orbitales no vinculantes. Todavía se describen de la manera LCAO-MO, pero sus\(\chi_{m,i}\) coeficientes no contienen contribuciones dominantes de más de un centro atómico.
Por último, existe un tipo de orbital que poseen todas las moléculas pero que se ignora en la mayoría de las discusiones elementales sobre la estructura electrónica. Todas las moléculas tienen los llamados orbitales de Rydberg. Estos orbitales pueden ser considerados como orbitales difusos grandes que describen las regiones del espacio que un electrón ocuparía si estuviera en presencia del catión molecular de caparazón cerrado correspondiente. Dos ejemplos de tales orbitales de Rydberg se muestran en la Figura 6.1.8. A la izquierda, vemos la órbita de Rydberg de\(NH_4\) y a la derecha, la de\(H_3N-CH_3\). La primera especie puede considerarse como un catión amónico de caparazón cerrado\(NH_4^+\) alrededor del cual reside una órbita de Rydberg. Esta última es metilamina protonada con su órbita de Rydberg.
