
7.3: Simulaciones de Dinámica Molecular
- Page ID
- 70887

Una cosa que el proceso MC no aborda directamente es la evolución temporal del sistema. Es decir, los pasos que uno examina en el algoritmo MC no son sencillos de asociar con una duración de tiempo, por lo que no está diseñado para calcular las tasas a las que ocurren los eventos. Si uno está interesado en simular tales procesos dinámicos, incluso cuando el sistema de moléculas N está en equilibrio o cerca de él, es más apropiado llevar a cabo una simulación clásica de dinámica molecular (MD). En dicho cálculo MD, se tienen que asignar valores iniciales para cada una de las coordenadas internas y externas de cada una de las\(N\) moléculas y un valor inicial de la energía cinética o impulso para cada coordenada, después de lo cual un algoritmo de propagación en el tiempo genera valores para las coordenadas y momentos en momentos posteriores. Por ejemplo, las coordenadas iniciales podrían elegirse cerca de las de un mínimo local en la superficie de energía y a los momentos iniciales asociados a cada coordenada se les podrían asignar valores elegidos de una distribución Maxwell-Boltzmann característica de una temperatura específica T. En tales casos, es común que luego permitir que la trayectoria MD se propague durante un período de tiempo lo suficientemente\(\Delta t\) largo como para permitir un mayor equilibrio de la energía entre todos los grados de libertad antes de extraer cualquier dato numérico para usar en la evaluación de valores promedio o crear histogramas de distancia entre partículas, por ejemplo.
Por lo general, no se elige solo un conjunto de tales coordenadas iniciales y momentos para generar una sola trayectoria. Más bien, se crea un conjunto de coordenadas iniciales y momentos diseñados para representar las condiciones experimentales que el cálculo MD va a simular. Luego se sigue la evolución temporal del sistema para cada conjunto de condiciones iniciales usando MD y se monitorean diversos resultados (por ejemplo, eventos reactivos, cruces de barrera, eventos de plegado o despliegue, ocurridos de quimiosorción, etc.) a lo largo de cada simulación de MD. Luego se utiliza un promedio sobre el conjunto de trayectorias para calcular promedios y crear histogramas para la simulación MD. El propósito de esta Sección es describir cómo se utiliza la MD para seguir la evolución del tiempo para tales simulaciones.
Propagación de trayectoria
Con cada coordenada teniendo su velocidad inicial\(\left(\dfrac{dq}{\delta t}\right)_0\) y su valor inicial\(q_0\) especificado, entonces se utilizan las ecuaciones de Newton escritas para un paso de tiempo de duración\(\delta t\) para propagarse\(q\) y\(dq/dt\) reenviar en el tiempo de acuerdo, por ejemplo, con la siguiente propagación de primer orden fórmula:
\[q(t+\delta t) = q_0 + \left(\dfrac{dq}{\delta t}\right)_0 dt\]
\[\dfrac{dq}{dt} (t+\delta t) = \left(\dfrac{dq}{dt}\right)_0 - \delta t \left[\left( \dfrac{∂V}{∂q} \right)_0\dfrac{1}{m_q}\right].\]
Aquí m_q es el factor de masa que conecta la velocidad\(dq/dt\) y el momento pq conjugado a la coordenada q:
\[p_q = m_q \dfrac{dq}{dt},\]
y\(-(∂V/∂q)_0\) es la fuerza a lo largo de la coordenada\(q\) en la geometría anterior\(q_0\). En la mayoría de las simulaciones de MD modernas, se pueden utilizar métodos numéricos más sofisticados para propagar las coordenadas y los momentos. Por ejemplo, el algoritmo Verlet ampliamente utilizado se deriva de la siguiente manera.
- Se expande el valor de la coordenada\(q\) en los pasos\(n+1^{\rm st}\) y de\(n-1^{\rm st}\) tiempo en la serie Taylor en términos de valores en el\(n\) primer paso de tiempo\[q_{n+1}=q_n+\left(\dfrac{dq}{dt}\right)_n\delta t +\dfrac{-\left( \dfrac{∂V}{∂q} \right)_n}{2m_q}\delta t^2-O(\delta t^3) \]
\[q_{n-1}=q_n-\left(\dfrac{dq}{dt}\right)_n\delta t +\dfrac{\left( \dfrac{∂V}{∂q} \right)_n}{2m_q}\delta t^2+O(\delta t^3)\] - Se agregan estas dos expansiones para obtener
\[q_{n+1}=2 q_n-q_{n-1}+\dfrac{-\left( \dfrac{∂V}{∂q} \right)_n}{2m_q}\delta t^2-O(\delta t^4)\]
lo que permite computar\(q_{n+1}\) en términos de\(q_{n}\)\(q_{n-1}\) y y la fuerza en el\(n^{\rm th}\) paso, sin requerir conocimiento de velocidades. - Si se restan las dos expansiones de Taylor, se obtiene\[\left(\dfrac{dq}{dt}\right)_{n+1}-\dfrac{q_{n+1}-q_{n-1}}{2\delta t}+O(\delta t^2)\] como expresión para la velocidad en el paso de\(n+1^{\rm st}\) tiempo en términos de las coordenadas en los\(n-1^{\rm st}\) pasos\(n+1^{\rm st}\) y.
Hay muchos otros esquemas de propagación de este tipo que se pueden usar en MD; cada uno tiene fortalezas y debilidades. En la presente Sección, me enfocaré en describir la idea básica de cómo se realizan las simulaciones de MD dejando el tratamiento de detalles sobre esquemas de propagación a fuentes más avanzadas como Simulaciones por Computadora de Líquidos, M. P. Allen y D. J. Tildesley, Oxford U. Press, Nueva York (1997).
Las fuerzas\(-(∂V/∂q)\) que aparecen en los algoritmos de propagación MD se pueden obtener como gradientes de una superficie de energía electrónica Born-Oppenheimer si esto es computacionalmente factible. Seguir este camino implica realizar lo que se llama MD de dinámica directa. Alternativamente, las fuerzas pueden calcularse a partir de derivadas de un campo de fuerza empírico. En este último caso, la energía potencial del sistema\(V\) se expresa en términos de funciones analíticas de
- longitudes de enlace intramoleculares, ángulos de unión y ángulos de torsión, así como
- distancias y orientaciones intermoleculares.
Los parámetros que aparecen en dichos campos de fuerza generalmente se han determinado a partir de cálculos de estructura electrónica en fragmentos moleculares, determinación espectroscópica de constantes de fuerza vibratoria y mediciones experimentales de fuerzas intermoleculares.
Campos de Fuerza
Interrumpamos nuestra discusión sobre la propagación MD de coordenadas y velocidades para examinar los ingredientes que suelen aparecer en los campos de fuerza mencionados anteriormente. En la Figura 7.3 c, vemos una molécula en la que se introducen diversas interacciones intramoleculares e intermoleculares.

El potencial total de un sistema que contiene una o más de tales moléculas en presencia de un disolvente (por ejemplo, agua) se escribe típicamente como una suma de potenciales intramoleculares (uno para cada molécula en el sistema) y potenciales itermoleculares. Los primeros generalmente se descomponen en una suma de interacciones covalentes que describen cómo varía la energía con el estiramiento del enlace, la flexión del enlace y la distorsión del ángulo diedro como se representa en la Figura 7.3 d.
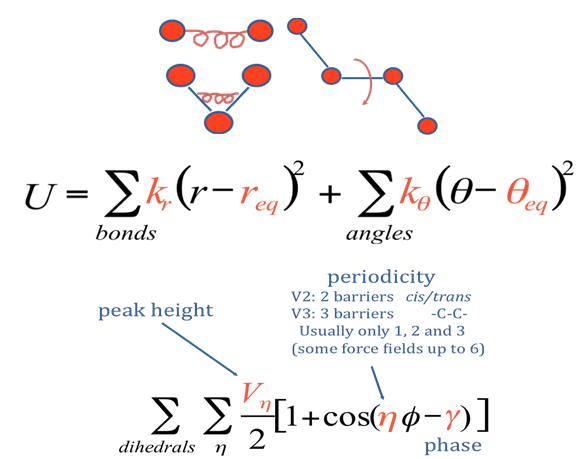
y no covalentes que describen interacciones electrostáticas y de van der Waals entre los átomos en la molécula a
\[V_{\rm noncovalent}=\sum_{i<j}^{\rm atoms} \left[\dfrac{A_{i,j}}{r_{i,j}^{12}}-\dfrac{B_{i,j}}{r_{i,j}^{6}}+\dfrac{q_iq_j}{\varepsilon r_{i,j}}\right].\]
Estas formas funcionales serían utilizadas para describir cómo\(V(q)\) cambia la energía con las longitudes de enlace (\(r\)) y ángulos (\(\theta,\phi\)) dentro de, por ejemplo, cada una de las moléculas que se muestran en la Figura 7. 3 c (llamémoslas moléculas de soluto) así como para cualquier molécula de agua que pueda estar presente (si estas ej., las moléculas se incluyen explícitamente en la simulación MD).
Las interacciones entre los moleulues de soluto y disolvente también se expresan a menudo en una forma que involucra interaciones electrostáticas y de van der Waals entre pares de átomos, uno en una molécula (soluto o disolvente) y el otro en otra molécula (soluto o disolvente)
\[V_{\rm intermolecular} = \sum_{i<j}^{\rm atoms} \left[\dfrac{A_{i,j}}{r_{i,j}^{12}}-\dfrac{B_{i,j}}{r_{i,j}^{6}}+\dfrac{q_iq_j}{\varepsilon r_{i,j}}\right].\]
Las fuerzas cartesianas sobre cualquier átomo dentro de una molécula de soluto o disolvente se calculan entonces para su uso en la simulación MD usando la regla de cadena para relacionar derivados con respecto a coordenadas cartesianas con derivados de los potenciales intramoleculares e intermoleculares anteriores con respecto a las distancias interatómicas y los ángulos que aparecen en ellos.
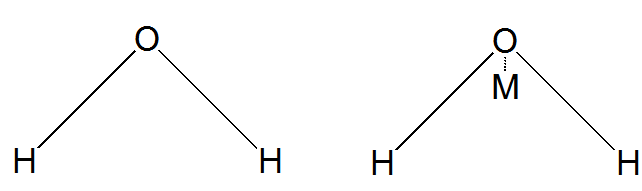
Debido a que el agua es un componente tan ubicuo en la química de fase condensada, se ha dedicado mucho esfuerzo a generar potenciales intermoleculares de alta precisión para describir las interacciones entre las moléculas de agua. En los populares modelos TIP3P y TIP4P, la interacción agua-agua viene dada por
\[V = \dfrac{A}{r_{OO}^{12}}-\dfrac{B}{r_{OO}^{6}}+\sum_{i,j}\dfrac{kq_iq_j}{r_{i,j}}.\]
donde RoO es la distancia entre los átomos de oxígeno de las dos moléculas de agua en Å, y los índices\(i\) y se\(j\) extienden sobre 3 o 4 sitios, respectivamente, para TIP3P o TIP4P, con sitios de\(i\) marcaje en una molécula de agua y sitios de\(j\) marcaje en la segunda molécula de agua. El parámetro\(k\) es 332.1 Å kcal mol -1. A y B son parámetros convencionales de Lennard-Jones para átomos de oxígeno y qi es la magnitud de la carga parcial en el sitio i-ésimo. En la Figura 7.3 d, mostramos cómo se definen los 3 o 4 sitios para estos dos modelos.
Los valores típicos para los parámetros se dan en la siguiente tabla.
|
r OH (Å) |
Grados de ángulo HOH |
r OM (Å) |
A (Å 12 kcal/mol) |
B (Å 6 kcal/mol) |
q O o q M |
q H |
|
|---|---|---|---|---|---|---|---|
|
TIP3P |
0.9572 |
104.52 |
582 x10 3 |
595 |
-0.834 |
0.417 |
|
|
TIP4P |
0.9672 |
104.52 |
0.15 |
600 x10 3 |
610 |
-1.04 |
0.52 |
En el modelo TIP3P, los tres sitios residen en los centros de oxígeno y dos de hidrógeno. Para TIP4P, el cuarto sitio se denomina sitio M y reside fuera del centro de oxígeno a una distancia de 0.15 a lo largo de la bisectriz de los dos enlaces O-H como se muestra en la Figura 7.3 d. Al usar el modelo TIP3P o TIP4P, las longitudes y ángulos del enlace intramolecular son a menudo limitado a permanecer fijo; al hacerlo, se dice que uno usa un modelo de agua rígida.
Existen variantes a estos dos modelos de 3 sitios y 4 sitios que, por ejemplo, incluyen interacciones de van der Waals entre\(H\) átomos en diferentes moléculas de agua, y hay modelos que incluyen más de 4 sitios, y modelos que permiten la polarización de cada molécula de agua inducida por los campos dipolares ( representado por las cargas parciales) de las otras moléculas de agua y de moléculas de soluto. Cuanto más detalle y complejidad se introduce, más esfuerzo computacional se necesita para realizar simulaciones de MD. En particular, las moléculas de agua que permiten la polarización son considerablemente más exigentes computacionalmente porque a menudo implican resolver de manera autoconsistente para la polarización de cada molécula por los potenciales de carga y dipolo de todas las demás moléculas, con cada potencial dipolo incluyendo tanto el dipolos permanentes e inducidos de esa molécula. El profesor John Wampler ha creado una página web en la que se resumen los detalles sobre los campos de fuerza de mecánica molecular introducidos anteriormente. Esta página web proporciona enlaces a numerosos paquetes de software que utilizan este tipo de campos de fuerza para realizar simulaciones MD. Estos enlaces también ofrecen información más detallada sobre el desempeño de diversos campos de fuerza, así como dar valores para los parámetros utilizados en esos campos de fuerza.
Los valores de los parámetros generalmente se obtienen mediante
- ajustar la forma funcional intramolecular o intermolecular (por ejemplo, como se muestra anteriormente) a energías obtenidas en cálculos de estructura electrónica en un gran número de geometrías, o
- ajustándolos para provocar simulaciones MD o MC que emplean el campo de fuerza para reproducir ciertas propiedades termodinámicas (por ejemplo, funciones de distribución radial, energías de solvatación, energías de vaporización, constantes de difusión), o alguna combinación de ambas. Es importante observar que el tipo de campos de fuerza discutidos anteriormente tienen limitaciones más allá de cuestiones de precisión. En particular, no están diseñados para permitir la ruptura de enlaces y la formación de enlaces, y representan la energía Born-Oppenheimer de un estado electrónico (la mayoría de las veces el suelo). Hay campos de fuerza diseñados explícitamente para incluir cambios de unión química, pero la mayoría de los paquetes MD no los incluyen. Cuando uno está interesado en tratar un problema que implica transiciones de un estado electrónico a otro (por ejemplo, en espectroscopia o cuando el sistema se somete a un salto superficial cerca de una intersección cónica), lo más común es utilizar un enfoque QM-MM combinado como hablamos en la Sección 6.1.3 del Capítulo 6. Un tratamiento QM de la porción del sistema que se somete a la transición electrónica se combina con un tratamiento de campo de fuerza (MM) del resto del sistema para llevar a cabo la simulación MD. Volvamos ahora al tema de la propagación de trayectorias dado un campo de fuerza y un conjunto de condiciones iniciales apropiadas para describir el sistema a simular.
Al aplicar uno de los algoritmos de propagación en el tiempo a todas las coordenadas y momentos de las\(N\) moléculas en el tiempo t, se genera un conjunto de nuevas coordenadas\(q(t+\delta t)\) y nuevas velocidades\(dq/dt(t+\delta t)\) apropiadas al sistema en el momento\(t+dt\). Usando estas nuevas coordenadas y momentos como\(q_0\)\((dq/\delta t)_0\) y evaluando las fuerzas\(–(∂V/∂q)_0\) en estas nuevas coordenadas, se pueden usar nuevamente las ecuaciones de propagación para generar otro conjunto de nuevas coordenadas y velocidades de tiempo finito. A través de la aplicación secuencial de este proceso, se genera una secuencia de coordenadas y velocidades que simulan el comportamiento del sistema. Al seguir estas coordenadas y momentos, se puede interrogar cualquier propiedad dinámica que le interese. Por ejemplo, se podrían monitorear distancias oxígeno-oxígeno a lo largo de una simulación MD de agua líquida con condiciones iniciales elegidas para representar el agua a una temperatura dada (T determinaría el momento inicial) para generar un histograma de distancias O-O. Esto permitiría construir el tipo de función de distribución radial que se muestra en la Figura 7. 3 usando simulación MD en lugar de MC. La función de distribución radial obtenida en dicha simulación MD debe ser idéntica a la obtenida de MC porque la mecánica estadística asume que el promedio de conjunto (MC) es igual al promedio a largo plazo (MD) de cualquier propiedad para un sistema en equilibrio. Por supuesto, también se podrían monitorear cantidades que dependen del tiempo, como la frecuencia con la que dos átomos de oxígeno se encuentran a cierta distancia, a lo largo de la simulación MD. Este tipo de interrogatorios no se pudo lograr usando MC porque no hay sentido del tiempo en las simulaciones MC.
En el Capítulo 8, nuevamente discuto el uso de la dinámica molecular clásica para seguir la evolución temporal de un sistema químico. Sin embargo, existe una diferencia fundamental entre el tipo de simulaciones descritas anteriormente y el caso que trato en el Capítulo 8. En el primero, uno permite que el sistema de moléculas N alcance el equilibrio (es decir, ya sea eligiendo cuidadosamente las coordenadas iniciales y los momentos o esperando hasta que la dinámica haya aleatorizado la energía) antes de monitorear la evolución temporal posterior. En el problema discutido en el Capítulo 8, utilizamos MD para seguir el progreso temporal de un sistema que representa una sola colisión bimolecular en dos haces cruzados de moléculas. Cada haz contiene moléculas cuyas velocidades de traducción iniciales están estrechamente definidas en lugar de Maxwell-Boltzmann distribuidas. En este caso, no permitimos que el sistema se equilibre porque no estamos tratando de modelar un sistema de equilibrio. En su lugar, seleccionamos un conjunto de condiciones iniciales que representan las moléculas en los dos haces y luego seguimos la dinámica de Newton para monitorear el resultado (por ejemplo, reacción o colisión no reactiva).
A diferencia del método MC, que es muy susceptible de computación paralela, las simulaciones MD son más difíciles de llevar a cabo de manera paralela. Ciertamente se pueden ejecutar muchas trayectorias clásicas diferentes en muchos nodos informáticos diferentes; sin embargo, distribuir una trayectoria sobre muchos nodos es difícil. La principal dificultad es que, por cada paso\(N\) de tiempo, todas las moléculas experimentan movimientos a nuevas coordenadas y momentos. Para calcular las fuerzas en todas las\(N\) moléculas se requiere del orden de\(N^2\) los cálculos (por ejemplo, cuando se utilizan potenciales aditivos por pares). En contraste, cada paso de MC requiere que se evalúe el cambio de energía potencial que acompaña al desplazamiento de una sola molécula. Esto usa solo del orden de los pasos\(N\) computacionales (nuevamente, para potenciales aditivos por pares).
Otro factor que complica las simulaciones MD tiene que ver con la amplia gama de escalas de tiempos que pueden estar involucradas. Por ejemplo, para que uno use un paso de tiempo dt lo suficientemente corto como para seguir movimientos de alta frecuencia (por ejemplo, estiramiento O-H) en una simulación de un ion o polímero en disolvente de agua, dt debe ser del orden de 10 -15 s. Para luego simular la difusión de un ion o el plegamiento de un polímero en estado líquido, que podría requerir 10 -4 s o más, uno tendría que llevar a cabo 10 11 pasos MD. Esto probablemente haría que la simulación no fuera factible. En la siguiente tabla ilustramos la amplia gama de escalas de tiempo que caracterizan diversos eventos que uno podría querer simular usando alguna forma de MD, y damos una idea de lo que es práctico usando simulaciones de MD en el año 2010.
|
10 -15 -10 -14 s |
10 -12 s |
10 -9 s |
10 -6 s |
10 -3 s |
110 s |
|---|---|---|---|---|---|
|
Vibración de unión C-H, N-H, O-H |
Rotación de molécula pequeña |
Duración de tiempo accesible de forma rutinaria para la simulación atomística de MD |
Duración del tiempo para la simulación heroica de MD atomista |
Duración del tiempo alcanzable usando técnicas de grano grueso a |
Tiempo necesario para el plegamiento de proteínas |
a. Estas técnicas se discuten en la Sección 7.3.3.
Debido a que uno no puede permitirse el lujo de realizar simulaciones que cubren 10 -3 -100 s usando pasos de tiempo necesarios para seguir las vibraciones de unión 10 -15 s, es necesario idear estrategias para enfocarse en movimientos cuyo marco de tiempo es de interés primordial mientras se ignoran o aproximan movimientos más rápidos. Por ejemplo, al realizar simulaciones de MD de larga duración, se pueden ignorar los movimientos intramoleculares de alta frecuencia simplemente no incluyendo estas coordenadas y momentos en la dinámica netwoniana (por ejemplo, como se hace cuando se usa un modelo rígido-agua discutido anteriormente). En otras palabras, uno simplemente congela ciertas longitudes y ángulos de unión. Por supuesto, se trata de una aproximación cuyas consecuencias deben ser probadas y justificadas, y ciertamente no sería un paso sabio a dar si esas coordenadas jugaran un papel clave en el proceso dinámico que se está simulando. Otro enfoque, llamado grano grueso implica reemplazar la descripción completamente atomística de componentes seleccionados del sistema por una descripción muy simplificada que implica significativamente menos coordenadas espaciales y momentos.
Graneado Grueso
El objetivo del grano grueso es llevar el costo computacional de una simulación al reino de la realidad. Esto se hace reemplazando la descripción completamente atomística del sistema, en la que coordenadas suficientes para especificar las posiciones (y, en MD, las velocidades) de cada átomo, por una descripción en términos de menos grupos funcionales a menudo denominados “perlas”. Los modelos TIP4P y TIP3P para el potencial de interacción agua-agua discutidos anteriormente no son modelos de grano grueso porque contienen tantos (o más) centros como átomos. Un ejemplo de un modelo de grano grueso para la interacción agua-agua es proporcionado por el modelo Stillinger-Weber (que se introdujo originalmente para tratar el Si tetraédrico) del agua introducida en V. Molinero y E. B. Moore, J. Phys. Chem. B 2009, 113, 4008—4016. Aquí, cada molécula de agua se describe solo por la ubicación de su núcleo de oxígeno (marcado ri para la i-ésima molécula de agua), y el potencial de interacción se da como una suma de términos de dos cuerpos y tres cuerpos
\[V=\sum_{i<j=1}^N A\varepsilon\left[B\left(\dfrac{\sigma}{r_{i,j}}\right)^p-\left(\dfrac{\sigma}{r_{i,j}}\right)^q\right]\exp\left(\dfrac{\sigma}{r_{i,j}-a\sigma}\right)\\+\sum_{i<j<k=1}^N \lambda \varepsilon[\cos\theta_{i,j,k}-\cos\theta_0]^2\exp\left(\dfrac{\gamma\sigma}{r_{i,j}-a\sigma}\right)\exp\left(\dfrac{\gamma\sigma}{r_{i,k}-a\sigma}\right)\]
donde\(r_{i,j}\) está la distancia entre el i-ésimo y el jésimo átomo de oxígeno,\(\theta_0 =\) 109.47 grados, y\(q_{i,j,k}\) es el ángulo entre el i-ésimo (en el centro), jésimo y késimo átomo de oxígeno. Los parámetros\(A\),\(B\),\(\varepsilon\)\(\sigma\), y\(a\) se utilizan para caracterizar diversas características del potencial; se necesitan diferentes valores para describir el comportamiento de Si, Ge, diamante o agua aunque todos puedan adoptar coordinación tetraédrica. La forma de la parte de tres cuerpos de este potencial está diseñada para guiar las orientaciones entre los átomos de oxígeno para adoptar el carácter tetraédrico.
Si bien el potencial anterior parece más complicado que, por ejemplo, la forma utilizada en el potencial TIP3P o TIP4P, tiene tres ventajas importantes a la hora de realizar simulaciones MD:
- Debido a que el potencial SW no contiene términos que varíen con la distancia como\(r^{-1}\) (es decir, no hay interacciones de Coulomb entre cargas parciales), es de rango cualitativamente más corto que los otros dos potenciales. Esto permite utilizar los cortes espaciales (es decir, ignorar las interacciones más allá de distancias mucho más cortas) de manera eficiente.
- 2. Para un sistema que contiene moléculas de\(N\) agua, los modelos TIP3P o TIP4P requieren uno para evaluar las funciones de las distancias entre\((3N)^2/2\) o\((4N)^2/2\) centros, mientras que el componente de dos cuerpos del SW implica solo\(N^2/2\) interacciones y el componente de tres cuerpos solo necesita ser evaluado para moléculas \(j\)y\(k\) que están cerca de la molécula\(i\).
- 3. Si, para los modelos atomísticos, se desea tratar los movimientos de estiramiento O-H y flexión H-O-H, se deben emplear pasos de tiempo MD de aproximadamente 10 -15 s. Para el modelo SW, los movimientos más rápidos implican movimientos relativos de los centros de oxígeno, los cuales ocurren en escalas de tiempo ca. 10 veces más largas. Esto significa que uno puede usar pasos MD más largos.
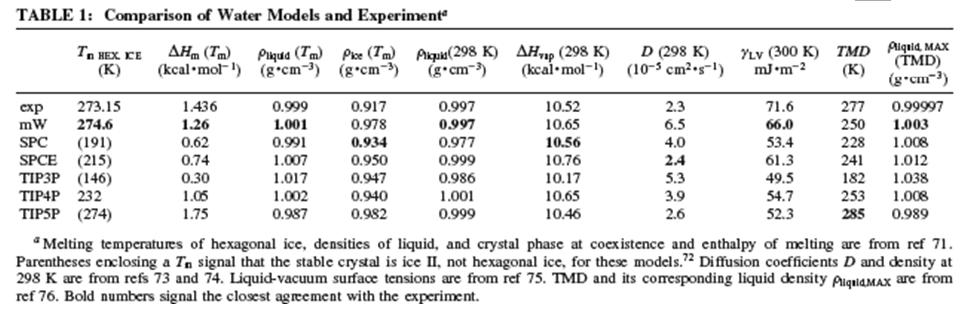
El resultado neto es que este modelo de grano grueso de la interacción agua-agua permite realizar simulaciones de MD para duraciones cualitativamente más largas. Por supuesto, esto solo es una ventaja si las simulaciones proporcionan resultados precisos. En la Tabla que se muestra a continuación (tomada de la referencia anterior), vemos resultados de simulación MD (así como resultados experimentales) obtenidos con el modelo anterior (mW), con varios modelos TiPnP, y con otros dos potenciales populares de agua-agua (SPC y SPCE) de los cuales está claro que el modelo mW de grano grueso es capaz de producir resultados confiables en una gama de propiedades termodinámicas.
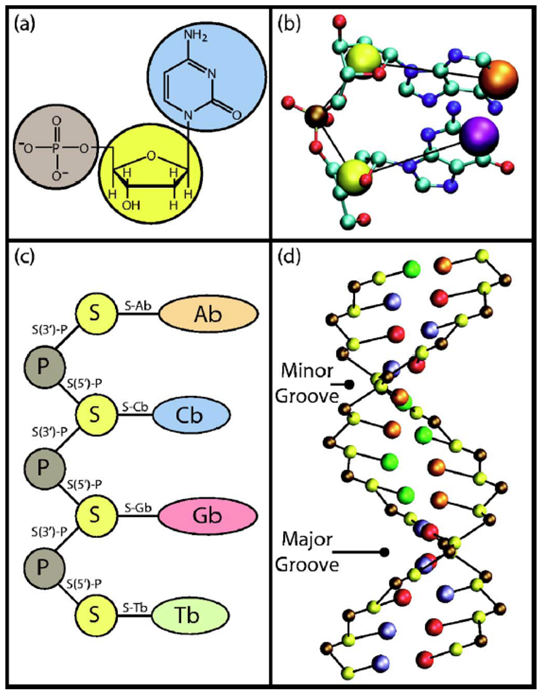
En la Tabla que se muestra a continuación, la referencia citada anteriormente especifica las localizaciones y masas de las perlas de fosfato, azúcar y base en forma B de la hélice de ADN. Las masas deben elegirse para que los movimientos dinámicos de grano grueso de estas unidades repliquen dentro de tolerancias razonables los movimientos del centro de masa de los grupos fosfato, azúcar y base cuando se realizan simulaciones atomísticas de MD en sistemas de prueba más pequeños que contienen estas unidades nucleotídicas.
El potencial\(V\) utilizado para realizar las simulaciones de MD de grano grueso viene dado por las ecuaciones que se muestran a continuación tomadas de la referencia anterior. Además de los términos habituales de estiramiento de enlace, flexión y diedro (n.b., ahora los enlaces se relacionan con enlaces entre cuentas más que entre átomos) que son similares a lo que vimos anteriormente en nuestra discusión sobre los campos de fuerza, existen términos adicionales.
- \(V_{\rm stack}\)describe las interacciones entre pares de bases apiladas p,
- \(V_{\rm bp}\)describe las interacciones de enlaces de hidrógeno entre bases, y
- \(V_{\rm ex}\)describe los efectos de volumen excluido.
\[V_{\rm total}=V_{\rm bond}+V_{\rm angle}+V_{\rm dihedral}+V_{\rm stack}+V_{\rm bp}+V_{\rm ex}+V_{\rm qq},\]
donde
\[V_{\rm bond}=\sum_i^{N_{\rm bond}}[k_1(d_i-d_{0_i})^2+k_2(d_i-d_{0_i})^4],\]
\[V_{\rm angle}=\sum_i^{N_{\rm angle}}\dfrac{k_\theta}{2}(\theta_i-\theta_{0_i})^2,\]
\[V_{\rm dihedral}=\sum_i^{N_{\rm dihedral}}k_\phi [1-\cos(\phi_i-\phi_{0_i})],\]
\[V_{\rm stack}=\sum_{i<j}^{N_{\rm st}} 4\varepsilon\left[\left(\dfrac{\sigma_{ij}}{r_{ij}}\right)^{12}-\left(\dfrac{\sigma_{ij}}{r_{ij}}\right)^6\right],\]
\[V_{\rm bp}=\sum_{\rm base pairs}^{N_{\rm bp}} 4\varepsilon_{\text{bp}{}_i}\left[5\left(\dfrac{\sigma_{\text{bp}{}_i}}{r_{ij}}\right)^{12}-6\left(\dfrac{\sigma_{\text{bp}{}_i}}{r_{ij}}\right)^{10}\right],\]
\ [V_ {\ rm ex} =\ suma_ {i<j} ^ {N_ {\ rm ex}}\ izquierda\ {\ begin {array} {cc}
4\ varepsilon\ izquierda [\ izquierda (\ dfrac {\ sigma_ {ij}} {r_ {ij}}\ derecha) ^ {12} -\ izquierda (\ dfrac {\ sigma_ _ {ij}} {r_ {ij}}\ derecha) ^6\ derecha] +\ varepsilon &\ texto {si} r_ {ij} <d_ {\ rm corte}\\
0 &\ texto {si} r_ {ij}\ ge d_ {\ rm corte}
\ end {array}\ derecho.,\]
\[V_{\rm qq}=\sum_{i<j}^N\dfrac{q_iq_j}{4\pi\varepsilon_0 \varepsilon_k r_{ij}}e^{-r'_{ij}\kappa_D}.\]
4. \(V_{\rm qq}\)es las interacciones coulómbicas seleccionadas entre unidades de fosfato, con su constante de decaimiento exponencial\(\kappa_D\) dada en términos de la llamada longitud de cribado Debye como se detalla en la referencia anterior.
Los valores de los parámetros utilizados en este potencial de campo de fuerza dados en la referencia anterior se reproducen en las dos Tablas que se muestran a continuación.
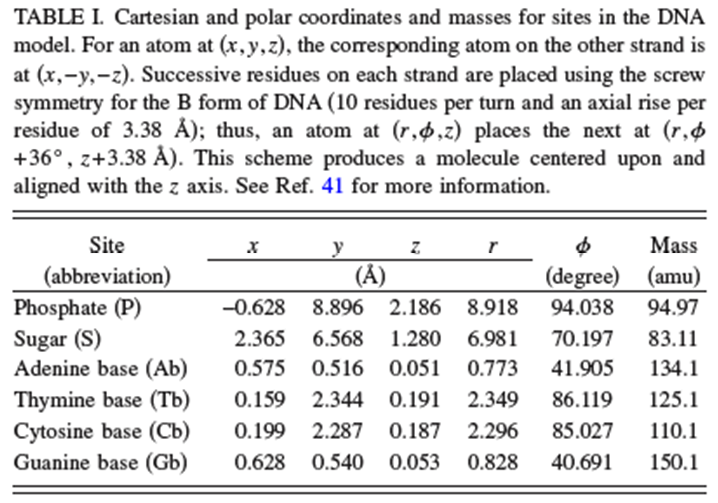
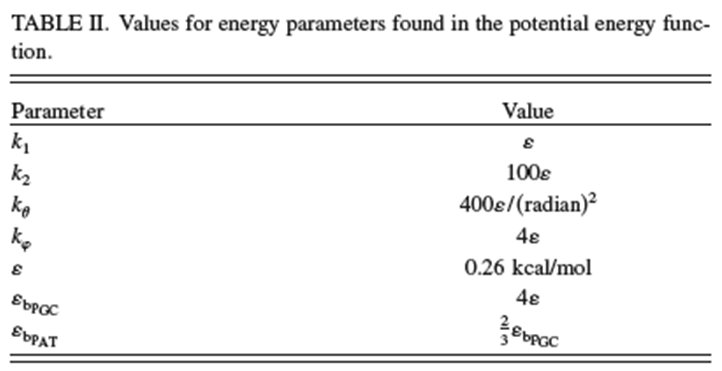
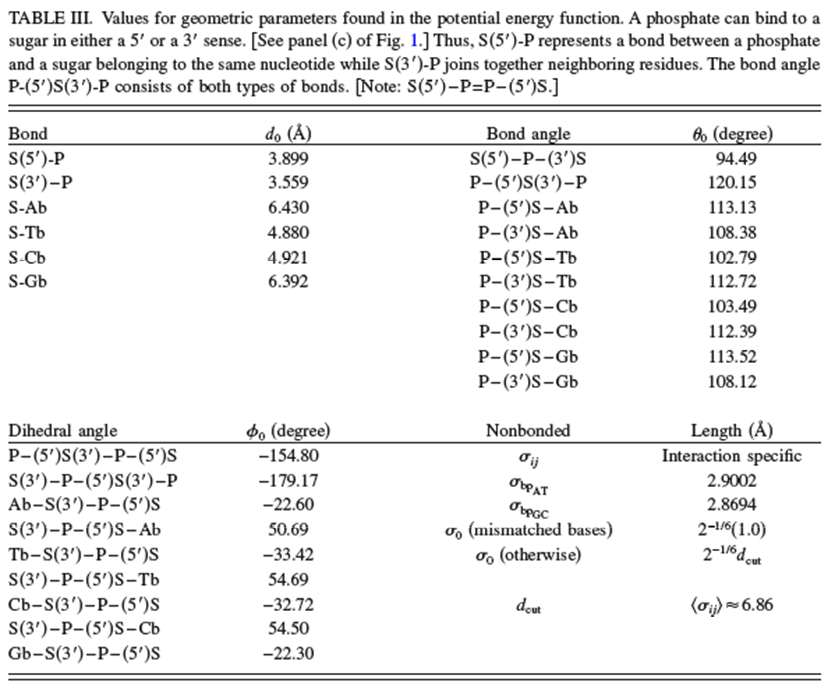
Aunque existen numerosos parámetros en este potencial, la clave del éxito de este grano grueso es que solo hay seis tipos de sitios cuyas posiciones y velocidades deben propagarse en la simulación MD: sitios de fosfato, sitios de azúcar y cuatro tipos de sitios base. Se trata de muchas menos coordenadas que surgirían en una simulación MD completamente atomística. Voy a referir al lector a la referencia citada anteriormente para obtener detalles sobre qué tan exitoso fue el grano grueso en este caso, pero no voy a profundizar en ello en este momento. Creo que los dos ejemplos que discutimos en esta Sección bastan para introducir el tema del grano grueso a los lectores de este texto.En resumen para esta Sección, las simulaciones clásicas MD no son difíciles de implementar si se tiene disponible una representación adecuada de lo intramolecular e intermolecular energía potencial V. Dichos cálculos se llevan a cabo rutinariamente en grandes biomoléculas o sistemas de medios condensados que contienen de miles a millones de centros atómicos. Sin embargo, existen dificultades relacionadas principalmente con las escalas de tiempo sobre las cuales cambian los movimientos moleculares y sobre las cuales el proceso que se simula, limitan el éxito de este método y que a menudo requieren emplear representaciones reducidas del sistema como en el grano grueso. En contraste, las simulaciones cuánticas de MD como las que describimos en la siguiente Sección son considerablemente más difíciles de llevar a cabo.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)