
14.2: La Primera Ley de la Termodinámica
- Page ID
- 70605

Asegúrese de comprender a fondo los siguientes conceptos esenciales:
- Definir: sistema, entorno, propiedades estatales, cambio de estado (cómo se calcula este último).
- Escribe la ecuación que define la Primera Ley, y explica su significado físico.
- Describir el significado de un camino en la termodinámica, y cómo se relaciona con las funciones estatales.
- Comentario sobre el significado del trabajo presión-volumen: ¿cómo se calcula? ¿Qué tipo de reacciones químicas implican el trabajo P-V?
- Definir procesos isotérmicos y adiabáticos, y dar ejemplos de cada uno.
- Utilizando como ejemplo la expansión de un gas, se establece la distinción fundamental entre cambios reversibles e irreversibles en términos del sistema + entorno.
- Explicar cómo el cambio de entalpía se relaciona con la energía interna, y cómo se puede observar experimentalmente.
- Definir la capacidad calorífica; explicar su significación física, y por qué hay una diferencia entre C p y C v.
“La energía no puede crearse ni destruirse” —esta ley fundamental de la naturaleza, más propiamente conocida como conservación de la energía, es familiar para cualquiera que haya estudiado ciencia. Bajo su nombre más formal de Primera Ley de la Termodinámica, rige todos los aspectos de la energía en las aplicaciones de la ciencia y la ingeniería. Es de especial importancia en la Química surge del hecho de que prácticamente todas las reacciones químicas van acompañadas de la captación o liberación de energía. Una de las cosas interesantes de la termodinámica es que aunque trata de la materia, no hace suposiciones sobre la naturaleza microscópica de esa materia. La termodinámica trata la materia en un sentido macroscópico; sería válida aunque la teoría atómica de la materia estuviera equivocada. Esta es una cualidad importante, porque significa que es poco probable que el razonamiento basado en la termodinámica requiera alteración a medida que salgan a la luz nuevos hechos sobre la estructura atómica y las interacciones atómicas.
Una visión termodinámica del mundo
En la termodinámica, debemos ser muy precisos en nuestro uso de ciertas palabras. Los dos más importantes de estos son el sistema y el entorno. Un sistema termodinámico es aquella parte del mundo a la que estamos dirigiendo nuestra atención. Todo lo que no forma parte del sistema constituye el entorno. El sistema y los alrededores están separados por un límite.
Si nuestro sistema es un mol de un gas en un contenedor, entonces el límite es simplemente la pared interna del contenedor mismo. El límite no necesita ser una barrera física; por ejemplo, si nuestro sistema es una fábrica o un bosque, entonces el límite puede estar donde queramos definirlo. Incluso podemos centrar nuestra atención en los iones disueltos en una solución acuosa de una sal, dejando las moléculas de agua como parte del entorno. La propiedad única que debe tener el límite es que esté claramente definida, por lo que podemos decir sin ambigüedades si una determinada parte del mundo está en nuestro sistema o en los alrededores.
Si la materia no puede atravesar el límite, entonces se dice que el sistema está cerrado; de lo contrario, está abierto. Un sistema cerrado aún puede intercambiar energía con el entorno a menos que el sistema sea aislado, en cuyo caso ni la materia ni la energía pueden atravesar el límite. El té en una botella Thermos cerrada se aproxima a un sistema cerrado en un corto intervalo de tiempo.
Las propiedades de un sistema son aquellas cantidades como la presión, el volumen, la temperatura y su composición, que en principio son medibles y capaces de asumir valores definidos. Por supuesto, existen muchas propiedades distintas a las mencionadas anteriormente; la densidad y la conductividad térmica son dos ejemplos. Sin embargo, la presión, el volumen y la temperatura tienen especial importancia porque determinan los valores de todas las demás propiedades; por lo tanto, se les conoce como propiedades de estado porque si se conocen sus valores entonces el sistema está en un estado definido.
Al tratar con la termodinámica, debemos ser capaces de definir inequívocamente el cambio en el estado de un sistema cuando éste se somete a algún proceso. Esto se hace especificando cambios en los valores de las diferentes propiedades de estado usando el símbolo Δ (delta) como se ilustra aquí para un cambio en el volumen:
\[ΔV = V_{final} – V_{initial} \label{1-1}\]
Podemos calcular valores delta similares para cambios en P, V, n i (el número de moles del componente i), y las otras propiedades estatales que conoceremos más adelante.
La energía interna es simplemente la totalidad de todas las formas de energía cinética y potencial del sistema. La termodinámica no hace distinción entre estas dos formas de energía y no asume la existencia de átomos y moléculas. Pero como estamos estudiando la termodinámica en el contexto de la química, podemos permitirnos apartarnos de la termodinámica “pura” lo suficiente como para señalar que la energía interna es la suma de la energía cinética del movimiento de las moléculas, y la energía potencial representada por los enlaces químicos entre los átomos y cualquier otra fuerza intermolecular que pueda ser operativa.
¿Cómo podemos saber cuánta energía interna posee un sistema? La respuesta es que no podemos, al menos no sobre una base absoluta; todas las escalas de energía son arbitrarias. Lo mejor que podemos hacer es medir los cambios en la energía. No obstante, somos perfectamente libres de definir la energía cero como la energía del sistema en algún estado de referencia arbitrario, para luego decir que la energía interna del sistema en cualquier otro estado es la diferencia entre las energías del sistema en estos dos estados diferentes.
La Primera Ley de la Termodinámica
Esta ley es uno de los principios más fundamentales del mundo físico. También conocida como Ley de Conservación de la Energía, establece que la energía no se puede crear ni destruir; sólo se puede redistribuir o cambiar de una forma a otra. Una forma de expresar esta ley que generalmente es más útil en Química es que cualquier cambio en la energía interna de un sistema viene dado por la suma del calor q que fluye a través de sus límites y el trabajo w realizado en el sistema por el entorno.
\[ΔU = q + w \label{2-1}\]
Esto dice que hay dos tipos de procesos, el calor y el trabajo, que pueden llevar a un cambio en la energía interna de un sistema. Dado que tanto el calor como el trabajo pueden medirse y cuantificarse, esto es lo mismo que decir que cualquier cambio en la energía de un sistema debe resultar en un cambio correspondiente en la energía del mundo fuera del sistema, es decir, la energía no puede crearse ni destruirse.
Hay una importante convención de señales para el calor y el trabajo que se espera que conozcas. Si el calor fluye hacia un sistema o el entorno para hacer trabajo en él, la energía interna aumenta y el signo de q o w es positivo. Por el contrario, el flujo de calor fuera del sistema o el trabajo realizado por el sistema será a expensas de la energía interna, y por lo tanto será negativo. (Tenga en cuenta que esto es lo contrario de la convención de signos que se utilizó comúnmente en gran parte de la literatura anterior a 1970). El significado completo de la Ecuación\(\ref{2-1}\) no puede ser captado sin entender que U es una función de estado. Esto significa que un cambio dado en la energía interna Δ U puede seguir una variedad infinita de caminos correspondientes a todas las combinaciones posibles de q y w que pueden sumar hasta un valor dado de Δ U.
Como ejemplo sencillo de cómo este principio puede simplificar nuestra comprensión del cambio, consideremos dos recipientes idénticos de agua inicialmente a la misma temperatura. Ponemos una llama debajo de una hasta que su temperatura haya subido 1°C y el agua del otro recipiente se agita vigorosamente hasta que su temperatura haya aumentado en la misma cantidad. Ahora no hay ninguna prueba física mediante la cual se pudiera determinar qué muestra de agua se calentó realizando trabajos sobre ella, permitiendo que el calor fluya hacia ella, o por alguna combinación de los dos procesos. Es decir, no hay base para decir que una muestra de agua ahora contiene más “trabajo”, y la otra más “calor”. Lo único que podemos saber con certeza es que ambas muestras han sufrido aumentos idénticos en la energía interna, y podemos determinar el valor de simplemente midiendo el aumento en la temperatura del agua.
Trabajo de presión-volumen
El tipo de trabajo más frecuentemente asociado al cambio químico ocurre cuando el volumen del sistema cambia debido a la desaparición o formación de sustancias gaseosas. Esto a veces se llama trabajo de expansión o trabajo fotovoltaico, y se puede entender más fácilmente por referencia a la forma más simple de materia con la que podemos tratar, el hipotético gas ideal.

La figura muestra una cantidad de gas confinado en un cilindro por medio de un pistón móvil. Los pesos colocados en la parte superior del pistón ejercen una fuerza f sobre el área de sección transversal A, produciendo una presión P = f/A que es contrarrestada exactamente por la presión del gas, de manera que el pistón permanece estacionario. Ahora supongamos que calentamos ligeramente el gas; según la ley de Charles, esto provocará que el gas se expanda, por lo que el pistón será forzado hacia arriba por una distancia Δ x. Dado que este movimiento es opuesto por la fuerza f, una cantidad de trabajo f Δ x será realizada por el gas en el pistón. Por convención, el trabajo realizado por el sistema (en este caso, el gas) en el entorno es negativo, por lo que el trabajo viene dado por
\[w = – f Δx \label{3-1}\]
Cuando se trata de un gas, es conveniente pensar en términos de las cantidades más relevantes de presión y volumen en lugar de fuerza y distancia. Podemos lograr esto multiplicando el segundo término por A/A que por supuesto lo deja sin cambios:
\[ w = -f Δx \dfrac{A}{A} \label{3-2}\]
Al agrupar los términos de manera diferente, pero aún sin cambiar nada, obtenemos
\[ w = -\dfrac{f}{A} Δx A \label{3-3}\]
Dado que la presión es fuerza por unidad de área y el producto de la longitud A y el área tiene las dimensiones de volumen, esta expresión se convierte en
\[w = –P ΔV \label{3-4}\]
Es importante señalar que aunque\(P\) y\(V\) son funciones de estado, el trabajo no lo es (por eso lo denotamos con una w minúscula). Como se muestra más adelante, la cantidad de trabajo realizado dependerá de si el mismo cambio de volumen neto se realiza en un solo paso (ajustando la presión externa a la presión final P), o en múltiples etapas ajustando la presión de restricción sobre el gas a valores sucesivamente más pequeños acercándose al valor final de\(P\).
Encuentra la cantidad de trabajo realizado en los alrededores cuando se permite que 1 litro de un gas ideal, inicialmente a una presión de 10 atm, se expanda a temperatura constante a 10 litros por:
- reducir la presión externa a 1 atm en un solo paso,
- reduciendo\(P\) primero a 5 atm, y luego a 1 atm,
- permitiendo que el gas se expanda a un espacio evacuado por lo que su volumen total es de 10 litros.
Solución
Primero, la nota th at\(ΔV\), que es una función de estado, es la misma para cada ruta:
\[V_2 = (10/1) × (1 L) = 10 L\]
por lo
\[ΔV = 9\; L\]
Para el camino (a)
w = — (1 atm) × (9 L) = —9 L-atm.
Para path (b), el trabajo se calcula para cada etapa por separado:
w = — (5 atm) × (2—1 L) — (1 atm) × (10—2 L) = —13 L-atm
Para la trayectoria (c) el proceso se llevaría a cabo retirando todos los pesos del pistón en la Figura\(\PageIndex{1}\) para que th en el gas se expanda a 10 L contra presión externa cero. En este caso w = (0 atm) × 9 L = 0; es decir, no se realiza ningún trabajo porque no hay fuerza para oponerse a la expansión.
Procesos adiabáticos e isotérmicos
Cuando un gas se expande, sí funciona en el entorno; la compresión de un gas a un volumen menor requiere de manera similar que el entorno realice trabajos sobre el gas. Si el gas se aísla térmicamente de los alrededores, entonces se dice que el proceso ocurre adiabáticamente. En un cambio adiabático, q = 0, por lo que la Primera Ley se convierte en Δ U = 0 + w. Dado que la temperatura del gas cambia con su energía interna, se deduce que la compresión adiabática de un gas provocará que se caliente, mientras que la expansión adiabática resultará en enfriamiento.
En contraste con esto, considere un gas que se deja escapar lentamente de un recipiente sumergido en un baño de temperatura constante. A medida que el gas se expande, sí funciona en el entorno y por lo tanto tiende a enfriarse, pero el gradiente térmico que resulta provoca que el calor pase al gas desde el entorno para compensar exactamente este cambio. A esto se le llama expansión isotérmica. En un proceso isotérmico la energía interna permanece constante y podemos escribir la Primera Ley (Ecuación\(Ref{2-1}\)) como
\[0 = q + w\]
o
\[q = –w\]
Esto ilustra que el flujo de calor y el trabajo realizado se equilibran exactamente entre sí.
Debido a que ningún aislamiento térmico es perfecto, no se producen procesos verdaderamente adiabáticos. Sin embargo, el flujo de calor lleva tiempo, por lo que una compresión o expansión que ocurre más rápidamente que el balance térmico puede considerarse adiabática para fines prácticos. Si alguna vez has usado una bomba manual para inflar una llanta de bicicleta, es posible que hayas notado que la parte inferior del cañón de la bomba puede calentarse bastante. Aunque una pequeña parte de este calentamiento puede deberse a la fricción, en su mayoría es el resultado del trabajo que usted (los alrededores) está haciendo en el sistema (el gas.)
La expansión adiabática y las contracciones son especialmente importantes para comprender el comportamiento de la atmósfera. Aunque comúnmente pensamos en la atmósfera como homogénea, realmente no lo es, debido en gran parte al calentamiento y enfriamiento desiguales sobre áreas localizadas. Debido a que la mezcla y la transferencia de calor entre paquetes de aire adyacentes no ocurren rápidamente, muchos fenómenos atmosféricos comunes pueden considerarse al menos cuasi-adiabáticos.
Procesos reversibles
De Ejemplo\(\PageIndex{1}\) vemos que cuando un gas se expande en vacío (\(P_{external} = 0\)el trabajo realizado es cero. Este es el trabajo mínimo que puede hacer el gas; ¿cuál es el trabajo máximo que el gas puede realizar en los alrededores? Para responder a esto, observe que se realiza más trabajo cuando el proceso se realiza en dos etapas que en una etapa; un simple cálculo mostrará que se puede obtener aún más trabajo al aumentar el número de etapas, es decir, al permitir que el gas se expanda contra una serie de presiones externas sucesivamente más bajas. Para extraer del proceso el máximo trabajo posible, la expansión tendría que realizarse en una secuencia infinita de pasos infinitesimales. Cada etapa produce un incremento de trabajo P Δ V que puede expresarse como (RT/V) dV e integrado:
\[ \begin{align} w &= \int_{V_1}^{V_2} \dfrac{RT}{V} dv \\[4pt] &= RT \ln \dfrac{V_2}{V_1} \label{3-5} \end{align}\]
Aunque tal camino (que corresponde a lo que se llama un proceso reversible) no se puede realizar en la práctica, se puede aproximar tan estrechamente como se desee.
Aunque ningún proceso real puede tener lugar de manera reversible (¡tomaría un tiempo infinitamente largo!) , los procesos reversibles juegan un papel esencial en la termodinámica. La razón principal de esto es que q rev y w rev son funciones estatales que son importantes y se calculan fácilmente. Además, muchos procesos reales se llevan a cabo de manera suficientemente gradual para que puedan ser tratados como procesos aproximadamente reversibles para facilitar el cálculo.
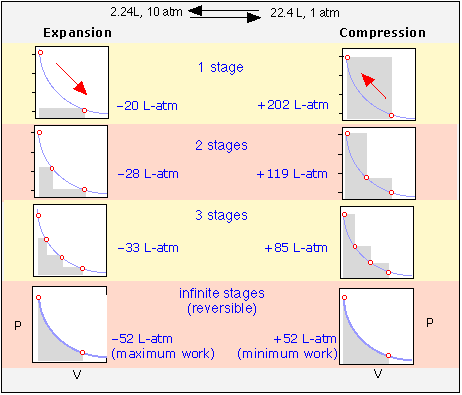
Cada ciclo de expansión-compresión deja el gas sin cambios, pero en todos menos en el de la fila inferior, los alrededores se alteran para siempre, habiendo dedicado más trabajo a comprimir el gas de lo que se realizó en él cuando el gas se expandió. Solo cuando los procesos se llevan a cabo en un número infinito de pasos se restaurará el sistema y el entorno a sus estados iniciales, esto es el significado de reversibilidad termodinámica.
Cambios de calor a presión constante: la entalpía
Para una reacción química que no realiza ningún trabajo en el entorno, el calor absorbido es el mismo que el cambio en la energía interna: q = Δ U. Pero muchos procesos químicos sí implican trabajo de una forma u otra:
- Si el volumen total de los productos de reacción excede al de los reactivos, entonces el proceso realiza trabajos sobre el entorno en la cantidad P Δ V, en la que P es la presión ejercida por el entorno (generalmente la atmósfera) sobre el sistema.
- Una reacción que impulsa una corriente eléctrica a través de un circuito externo realiza trabajos eléctricos en el entorno.
Para un proceso isotérmico, el trabajo presión-volumen afecta el calor q
Consideraremos solo el trabajo presión-volumen en esta lección. Si el proceso se lleva a cabo a una presión constante, entonces el trabajo viene dado por P Δ V y el cambio en la energía interna será
\[ΔU = q – PΔV \label{4-1}\]
Tenga en cuenta por qué\(q\) es tan importante: el flujo de calor dentro o fuera del sistema es directamente medible. Δ U, al ser “interno” al sistema, no es directamente observable. Así, la cantidad de calor que pasa entre el sistema y el entorno viene dada por
\[q = ΔU + PΔV \label{4-2}\]
Esto significa que si una reacción exotérmica va acompañada de un aumento neto de volumen bajo condiciones de presión constante, se debe absorber algo de calor adicional a Δ U para abastecer la energía gastada como trabajo realizado en el entorno si la temperatura ha de permanecer inalterada ( proceso isotérmico.)
Para la mayoría de los propósitos prácticos, los cambios en el volumen del sistema solo son significativos si la reacción va acompañada de una diferencia en los moles de reactivos gaseosos y productos.
Por ejemplo, en la reacción H 2 (g) + O 2 (g) → ½H 2 O (l), el volumen total del sistema disminuye de ese correponding a 2 moles de reactivos gaseosos a 0.5 mol de agua líquida que ocupa solo 9 mL — un volumen tan pequeño en comparación con la de los reactivos que se puede descuidar sin error significativo. Entonces, lo único que realmente nos preocupa es la diferencia en el número de moles de gas Δ n g:
Δ n g = (0 — 2) mol = —2 mol
Esto corresponde a una contracción neta (expansión negativa) del sistema, lo que significa que los alrededores realizan trabajos sobre el sistema.
El volumen molar de un gas ideal a 25° C y 1 atm es
(298/273) × (22.4 L mol —1) = 24.5 L mol —1
Recuerda la convención de signos: un flujo de calor o desempeño de trabajo que suministre energía es positivo; si consume energía, es negativo. Así, el trabajo realizado por el entorno disminuye la energía del entorno (w surr < 0) e incrementa la energía del sistema (w sys > 0).
y el trabajo realizado (por el entorno en el sistema) es
(1 atm) (—2 mol) (24.5 L mol —1) = —49.0 L-atm.
Utilizando el factor de conversión 1 J = 101.3 J, y teniendo en cuenta que el trabajo realizado en el sistema suministra energía al sistema, el trabajo asociado únicamente con el cambio de volumen del sistema incrementa su energía en
(101.3 J/L-atm) (—49.0 L-atm) = 4964 J = 4.06 kJ
La reacción anterior H 2 (g) + ½ O 2 (g) → H 2 O (l) se lleva a cabo a una presión constante de 1 atm y una temperatura constante de 25° C. ¿Qué cantidad de calor q cruzará el límite del sistema (y en qué dirección?) Para esta reacción, el cambio en la energía interna es Δ U = —281.8 kJ/mol.
Solución
281.8 kJ de calor. El trabajo realizado por el entorno suministra una energía adicional de P Δ V = 4.06 kJ al sistema. El cambio energético total del sistema es asíq = (—281.8 + 4.06) k J = —285.8 kJ
(Ec. 4.2 supra.) Para mantener la temperatura constante de 25°, una cantidad equivalente de calor debe pasar del sistema al entorno.
Esta terminología puede ser algo engañosa a menos que se tenga en cuenta que las condiciones Δ P y Δ T se refieren a las diferencias entre los estados inital y final del sistema —es decir, antes y después de la reacción. Durante el tiempo en que la reacción esté en curso, la temperatura de la mezcla subirá o bajará, dependiendo de si el proceso es exotérmico o endotérmico. Pero debido a que Δ T es una función de estado, su valor es independiente de lo que sucede “entre” el estado inicial (reactivos) y el estado final (productos). Lo mismo es cierto de Δ V.
¡La entalpía oculta el trabajo y lo ahorra también!
Debido a que la mayoría de los cambios químicos que tratamos tienen lugar a presión constante, sería tedioso tener que lidiar explícitamente con los detalles de trabajo presión-volumen que se describieron anteriormente. Afortunadamente, los químicos han encontrado una forma de evitar esto; simplemente han definido una nueva función de estado que incorpora y así esconde dentro de sí cualquier término relacionado con tipos incidentales de trabajo (P-V, eléctrico, etc.). Dado que tanto Δ P como Δ V en la Ecuación\(\ref{4-2}\) son funciones de estado\(q_P\), entonces, el calor que se absorbe o libera cuando un proceso tiene lugar a presión constante, también debe ser una función de estado y se conoce como el cambio de entalpía Δ H
\[ΔH ≡ q_P = ΔU + PΔV \label{4-3}\]
Dado que la mayoría de los procesos que ocurren en el laboratorio, en la superficie de la tierra, y en los organismos lo hacen bajo una presión constante de una atmósfera, la Ecuación\(\ref{4-3}\) es la forma de la Primera Ley que es de mayor interés para la mayoría de nosotros la mayor parte del tiempo.
El gas cloruro de hidrógeno se disuelve fácilmente en agua, liberando 75.3 kJ/mol de calor en el proceso. Si un mol de HCl a 298 K y 1 atm de presión ocupa 24.5 litros, encuentra Δ U para el sistema cuando un mol de HCl se disuelva en agua bajo estas condiciones.
Solución
En este proceso el volumen de líquido permanece prácticamente sin cambios, por lo que Δ V = —24.5 L. El trabajo realizado es
\[ \begin{align*} w &= –PΔV \\[4pt] &= –(1\; atm)(–24.5\; L) \\[4pt] &= 24.6 \;L-atm \end{align*}\]
(El trabajo es positivo porque se está haciendo en el sistema ya que su volumen disminuye debido a la disolución del gas en el volumen mucho menor de la solución). Usando el factor de conversión 1 L-atm = 101.33 J mol —1 y sustituyendo en la Ecuación\ ref {4-3} obtenemos
\[ \begin{align*} ΔU &= q +PΔV \\[4pt] = –(75,300\; J) + [101.33\; J/L-atm) (24.5\; L-atm)] \\[4pt] &= –72.82\; kJ \end{align*}\]
Es decir, si el HCl gaseoso pudiera disolverse sin cambio de volumen, el calor liberado por el proceso (75.3 kJ) haría que la energía interna del sistema disminuyera en 75.3 kJ. Pero la desaparición de la fase gaseosa reduce el volumen del sistema. Esto equivale a la compresión del sistema por la presión de la atmósfera realizando trabajos sobre él y consumiendo parte de la energía que de otro modo sería liberada, reduciendo el valor neto de Δ U a —72.82 k J.
La capacidad calorífica
Para los sistemas en los que no se produce ningún cambio en la composición (reacción química), las cosas son aún más simples: a una muy buena aproximación, la entalpía depende únicamente de la temperatura. Esto significa que la temperatura de dicho sistema puede servir como medida directa de su entalpía. La relación funcional entre la energía interna y la temperatura viene dada por la capacidad calorífica medida a presión constante:
\[ c_p =\dfrac{dH}{dT} \label{5-1}\]
(o Δ H /Δ T sobre una duración finita) Una cantidad análoga relaciona la capacidad calorífica a volumen constante con la energía interna:
\[ c_v =\dfrac{dU}{dT} \label{5-2}\]
Cuanto mayor sea la capacidad calorífica de una sustancia, menor será el efecto de una determinada absorción o pérdida de calor sobre su temperatura. La capacidad calorífica se puede expresar en julios o calorías por mol por grado (capacidad calorífica molar), o en julios o calorías por gramo por grado; esta última se llama la capacidad calorífica específica o simplemente el calor específico. La diferencia entre\(c_p\) y\(c_v\) es de importancia solo cuando el volumen del sistema cambia significativamente, es decir, cuando aparecen diferentes números de moles de gases a ambos lados de la ecuación química. Para reacciones que involucran solo líquidos y sólidos,\(c_p\) y\(c_v\) son para todos los fines prácticos idénticos.