
15.2: Reglas de Entropía
- Page ID
- 71037

Se espera que sea capaz de definir y explicar la significación de los términos identificados en negrita.
- Un proceso reversible es uno que se lleva a cabo en infinitosetapas simples después de las cuales, al deshacer, tanto el sistema como el entorno (es decir, el mundo) permanecen sin cambios (ver el ejemplo de expansión-compresión de gas a continuación). Aunque el verdadero cambio reversible no se puede realizar en la práctica, siempre se puede aproximar.
- ((en el que se lleva a cabo un proceso.
- A medida que un proceso se lleva a cabo de una manera más reversible, el valor de w se acerca a su valor máximo posible, y q se acerca a su valor mínimo posible.
- Aunque q no es una función de estado, el cociente q rev /T es, y se conoce como la entropía.
- energía dentro de un sistema.
- La entropía de una sustancia aumenta con su peso molecular y complejidad y con la temperatura. La entropía también aumenta a medida que la presión o concentración se vuelve más pequeña. Las entropías de los gases son mucho mayores que las de las fases condensadas.
- La entropía absoluta de una sustancia pura a una temperatura dada es la suma de toda la entropía que adquiriría al calentarse desde cero absoluto (donde S =0) hasta la temperatura particular.
La entropía es uno de los conceptos más fundamentales de la ciencia física, con consecuencias de largo alcance que van desde la cosmología hasta la química. También está ampliamente mal representado como una medida de “desorden”, como discutimos a continuación. El físico alemán Rudolf Clausius originó el concepto como “energía que se desperdició” a principios de la década de 1850, y su definición pasó por una serie de definiciones más precisas en los próximos 15 años.
Anteriormente, explicamos cómo la tendencia de la energía térmica a dispersarse lo más ampliamente posible es lo que impulsa todos los procesos espontáneos, incluyendo, por supuesto, las reacciones químicas. Ahora necesitamos entender cómo la dirección y el alcance de la difusión y distribución de la energía pueden relacionarse con las propiedades termodinámicas medibles de las sustancias, es decir, de los reactivos y productos.
Recordarás que cuando una cantidad de calor q fluye de un cuerpo más cálido a uno más frío, permitiendo que la energía térmica disponible se propague y poble más microestados, esa relación q/T mide la extensión de esta dispersión de energía. Resulta que también podemos generalizar esto a otros procesos, pero existe una dificultad con el uso de q porque no es una función de estado; es decir, su valor depende de la vía o manera en que se lleve a cabo un proceso. Esto significa, por supuesto, que el cociente q/T tampoco puede ser una función de estado, así que no podemos usarlo para obtener diferencias entre reactivos y productos como lo hacemos con las otras funciones de estado. La forma de evitar esto es restringir nuestra consideración a una clase especial de vías que se describen como reversibles.
Cambios reversibles e irreversibles
Se dice que un cambio ocurre reversiblemente cuando se puede llevar a cabo en una serie de pasos infinitesimales, cada uno de los cuales se puede deshacer haciendo un cambio igualmente minucioso a las condiciones que provocan el cambio. Por ejemplo, la expansión reversible de un gas se puede lograr reduciendo la presión externa en una serie de pasos infinitesimales; revertir cualquier paso restaurará el sistema y el entorno a su estado anterior. De igual manera, el calor puede transferirse reversiblemente entre dos cuerpos cambiando la diferencia de temperatura entre ellos en pasos infinitesimales cada uno de los cuales se puede deshacer invirtiendo la diferencia de temperatura.
El ejemplo más ampliamente citado de un cambio irreversible es la libre expansión de un gas en vacío. Si bien el sistema siempre se puede restaurar a su estado original recomprimiendo el gas, esto requeriría que los alrededores realicen trabajos sobre el gas. Dado que el gas no trabaja en el entorno en una expansión libre (la presión externa es cero, por lo que P Δ V = 0,) habrá un cambio permanente en el entorno. Otro ejemplo de cambio irreversible es la conversión del trabajo mecánico en calor por fricción; no hay manera, al invertir el movimiento de un peso a lo largo de una superficie, de que el calor liberado por fricción pueda ser restaurado al sistema.
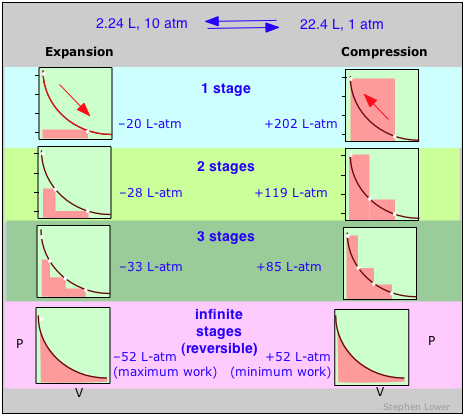
Estos diagramas muestran la misma expansión y compresión ±ΔV realizada en diferentes números de pasos que van desde un solo paso en la parte superior hasta un número “infinito” de pasos en la parte inferior. A medida que aumenta el número de pasos, los procesos se vuelven menos irreversibles; es decir, disminuye la diferencia entre el trabajo realizado en expansión y el requerido para recomprimir el gas. En el límite de un número “infinito” de pasos (abajo), estos términos de trabajo son idénticos, y tanto el sistema como el entorno (el “mundo”) no cambian por el ciclo de expansión-compresión. En todos los demás casos el sistema (el gas) se restablece a su estado inicial, pero los alrededores cambian para siempre.
Un cambio reversible es aquel que se lleva a cabo de tal manera que, al deshacer, tanto el sistema como el entorno (es decir, el mundo) permanecen sin cambios.
No hace falta decir, claro, que cualquier proceso que proceda en pasos infinitesimales tardaría infinitamente en ocurrir, por lo que la reversibilidad termodinámica es una idealización que nunca se logra en procesos reales, excepto cuando el sistema ya está en equilibrio, en cuyo caso no ocurrirá ningún cambio de todos modos ! Entonces, ¿por qué es tan importante el concepto de un proceso reversible?
La respuesta se puede ver recordando que el cambio en la energía interna que caracteriza a cualquier proceso puede distribuirse de infinidad de formas entre el flujo de calor a través de los límites del sistema y el trabajo realizado sobre o por el sistema, como lo expresa la Primera Ley Δ U = q + w. Cada combinación de q y w representa una ruta diferente entre los estados inicial y final. Se puede demostrar que como un proceso como la expansión de un gas se lleva a cabo en series sucesivamente más largas de etapas menores, el valor absoluto de q se acerca a un mínimo, y el de w se acerca a un máximo que es característico del proceso particular.
Así, cuando un proceso se lleva a cabo de manera reversible, el término w en la expresión de la Primera Ley tiene su mayor valor posible, y el término q está en su menor. Estas cantidades especiales w max y q min (que denotamos como q rev y pronunciamos “q-reversible”) tienen valores únicos para cualquier proceso dado y, por lo tanto, son funciones de estado.
Trabajo y reversibilidad
Para un proceso que intercambia reversiblemente una cantidad de calor q rev con el entorno, el cambio de entropía se define como
\[ \Delta S = \dfrac{q_{rev}}{T} \label{23.2.1}\]
Esta es la forma básica de evaluar Δ S para procesos de temperatura constante como los cambios de fase, o la expansión isotérmica de un gas. Para procesos en los que la temperatura no es constante como el calentamiento o enfriamiento de una sustancia, la ecuación debe integrarse sobre el rango de temperatura requerido, como se analiza a continuación.
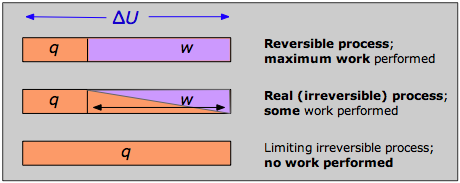
Si ningún proceso real puede tener lugar de manera reversible, ¿de qué sirve una expresión que involucra q rev? Este es un punto bastante fino que debes entender: aunque la transferencia de calor entre el sistema y el entorno es imposible de lograr de una manera verdaderamente reversible, este camino idealizado solo es crucial para la definición de Δ S; en virtud de ser una función de estado, el mismo valor de Δ S se aplicará cuando el sistema sufra el mismo cambio neto a través de cualquier vía. Por ejemplo, el cambio de entropía que sufre un gas cuando su volumen se duplica a temperatura constante será el mismo independientemente de que la expansión se lleve a cabo en 1000 pequeños pasos (tan reversible como sea probable que permita la paciencia) o por un solo paso (¡una vía tan irreversible como puedas conseguir!) expansión en un vacío.
El significado físico de la entropía
La entropía es una medida del grado de dispersión y distribución de la energía térmica dentro de un sistema. Esta “difusión y distribución” puede ser la difusión de la energía térmica en un mayor volumen de espacio o su distribución entre microestados previamente inaccesibles del sistema. En la siguiente tabla se muestra cómo se aplica este concepto a una serie de procesos comunes.
| sistema y proceso | fuente de aumento de entropía del sistema |
|---|---|
| Se baraja una baraja de cartas, o se lanzan al azar 100 monedas, inicialmente con la cabeza hacia arriba. | Esto no tiene nada que ver con la entropía porque los macroobjetos son incapaces de intercambiar energía térmica con el entorno dentro de la escala de tiempo del proceso |
| Dos bloques idénticos de cobre, uno a 20°C y el otro a 40°C, se ponen en contacto. | El bloque más frío contiene más microestados desocupados, por lo que el calor fluye desde el bloque más cálido hasta que se pueblan números iguales de microestados en los dos bloques. |
| Un gas se expande isotérmicamente al doble de su volumen inicial. | Una cantidad constante de energía térmica se extiende sobre un mayor volumen de espacio |
| 1 mol de agua se calienta a 1C°. | El aumento de la energía térmica hace que los microestados adicionales sean accesibles. (El incremento es por un factor de alrededor de 10 20,000,000,000,000, 000,000,000.) |
| Se permiten mezclar volúmenes iguales de dos gases. | El efecto es el mismo que permitir que cada gas se expanda al doble de su volumen; la energía térmica en cada uno ahora se extiende sobre un volumen mayor. |
| Un mol de dihidrógeno, H 2, se coloca en un recipiente y se calienta a 3000K. | Algunos de los H 2 se disocian a H porque a esta temperatura hay más microestados térmicamente accesibles en los 2 moles de H. |
| La mezcla de reacción anterior se enfría a 300K. | La composición vuelve a prácticamente todo el H 2 porque esta molécula contiene más microestados térmicamente accesibles a bajas temperaturas. |
La entropía es una cantidad extensa; es decir, es proporcional a la cantidad de materia en un sistema; así 100 g de cobre metálico tiene el doble de entropía de 50 g a la misma temperatura. Esto tiene sentido porque la pieza más grande de cobre contiene el doble de niveles de energía cuantificados capaces de contener la energía térmica.
Entropía y “desorden”
La entropía aún se describe, particularmente en los libros de texto más antiguos, como una medida del trastorno. En un sentido técnico estrecho esto es correcto, ya que la difusión y distribución de la energía térmica sí tiene el efecto de aleatorizar la disposición de la energía térmica dentro de un sistema. Pero simplemente equiparar entropía con “desorden” sin más calificación es extremadamente engañoso porque es demasiado fácil olvidar que la entropía (y la termodinámica en general) se aplica únicamente a sistemas de nivel molecular capaces de intercambiar energía térmica con el entorno. Llevar estos conceptos a macro sistemas puede producir analogías convincentes, pero ya no es ciencia. es mucho mejor evitar el término “desorden” por completo al discutir la entropía.

Entropía y Probabilidad
La distribución de la energía térmica en un sistema se caracteriza por el número de microestados cuantificados que son accesibles (es decir, entre los cuales se puede compartir la energía); mientras más de estos haya, mayor es la entropía del sistema. Esta es la base de una definición alternativa (y más fundamental) de entropía
\[\color{red} S = k \ln Ω \label{23.2.2}\]
en la que k es la constante de Boltzmann (la constante de gas por molécula, 1.38 10 —23 J K —1) y Ω (omega) es el número de microestados que corresponden a un macroestado dado del sistema. Cuantos más microestados de este tipo, mayor es la probabilidad de que el sistema esté en el macroestado correspondiente. Para cualquier macroestado físicamente realizable, la cantidad Ω es un número inimaginablemente grande, típicamente alrededor\(10^{10^{25}}\) de un mol. En comparación, el número de átomos que componen la tierra es de aproximadamente\(10^{50}\). Pero a pesar de que está más allá de la comprensión humana comparar números que parecen estar al borde del infinito, la energía térmica contenida en los sistemas físicos reales logra descubrir la mayor de estas cantidades sin ninguna dificultad, instalándose rápidamente en el macroestado más probable para un conjunto dado de condiciones.
La razón S depende del logaritmo de Ω es fácil de entender. Supongamos que tenemos dos sistemas (contenedores de gas, digamos) con S 1, Ω 1 y S 2, Ω 2. Si ahora redefinimos esto como un solo sistema (sin mezclar realmente los dos gases), entonces la entropía del nuevo sistema será
\[S = S_1 + S_2\]
pero el número de microestados será el producto Ω 1 Ω 2 ya que para cada estado del sistema 1, el sistema 2 puede estar en cualquiera de Ω 2 estados. Porque
\[\ln(Ω_1Ω_2) = \ln Ω_1 + \ln Ω_2\]
De ahí que se conserve la aditividad de la entropía.
Si alguien pudiera hacer una película mostrando los movimientos de átomos individuales de un gas o de un sistema de reacción química en su estado de equilibrio, no hay forma de que se pueda determinar, al verla, si la película se está reproduciendo en la dirección hacia adelante o hacia atrás. Los físicos describen esto diciendo que tales sistemas poseen simetría de inversión en el tiempo; ni la mecánica clásica ni cuántica ofrecen ninguna pista sobre la dirección del tiempo.
Sin embargo, cuando una película que muestra cambios en el nivel macro escópico se reproduce hacia atrás, la rareza es claramente evidente para cualquiera; si ves libros volando de una mesa o té siendo succionado de nuevo en una bolsa de té (o una reacción química corriendo a la inversa), inmediatamente sabrás que algo está mal. En este nivel, el tiempo claramente tiene una dirección, y muchas veces se nota que debido a que la entropía del mundo en su conjunto siempre aumenta y nunca disminuye, es la entropía la que le da al tiempo su dirección. Es por esta razón que la entropía a veces se denomina “flecha del tiempo”.
Pero aquí hay un problema: la termodinámica convencional es capaz de definir el cambio de entropía solo para procesos reversibles que, como sabemos, tardan infinitamente en realizarse. Entonces nos encontramos ante la aparente paradoja de que la termodinámica, que trata únicamente de las diferencias entre los estados y no de los viajes entre ellos, es incapaz de describir el proceso mismo de cambio por el cual somos conscientes del flujo del tiempo.
La dirección del tiempo es revelada al químico por el avance de una reacción hacia su estado de equilibrio; una vez alcanzado el equilibrio, cesa el cambio neto que conduce a él, y desde el punto de vista de ese sistema particular, el flujo del tiempo se detiene. Si extendemos la misma idea al sistema mucho más amplio del mundo en su conjunto, esto lleva al concepto de la “muerte por calor del universo” que se mencionó brevemente en la lección anterior.
Entropías absolutas
Los valores energéticos, como saben, son todos relativos, y deben definirse en una escala completamente arbitraria; no existe tal cosa como la energía absoluta de una sustancia, por lo que podemos definir arbitrariamente la entalpía o energía interna de un elemento en su forma más estable a 298K y 1 atm de presión como cero. Lo mismo no es cierto de la entropía; dado que la entropía es una medida de la “dilución” de la energía térmica, se deduce que cuanto menor sea la energía térmica disponible para propagarse a través de un sistema (es decir, cuanto menor sea la temperatura), menor será su entropía. Es decir, a medida que la temperatura absoluta de una sustancia se acerca a cero, también lo hace su entropía. Este principio es la base de la Tercera ley de la termodinámica, que establece que la entropía de un sólido perfectamente ordenado a 0 K es cero.
Tercera ley de la termodinámica
La entropía de un sólido perfectamente ordenado a 0 K es cero.
La entropía absoluta de una sustancia a cualquier temperatura superior a 0 K debe determinarse calculando los incrementos de calor q requeridos para llevar la sustancia de 0 K a la temperatura de interés, y luego sumando las proporciones q/T. Se necesitan dos tipos de mediciones experimentales:
- Las entalpías asociadas a cualquier cambio de fase que pueda sufrir la sustancia dentro del rango de temperatura de interés. La fusión de un sólido y la vaporización de un líquido corresponden a incrementos considerables en el número de microestados disponibles para aceptar energía térmica, por lo que a medida que ocurren estos procesos, la energía fluirá hacia un sistema, llenando estos nuevos microestados en la medida requerida para mantener una temperatura constante (la congelación o punto de ebullición); estas entradas de energía térmica corresponden a los calores de fusión y vaporización. El aumento de entropía asociado con la fusión, por ejemplo, es jus t ΔH fusión/T m.
- La capacidad calorífica C de una fase expresa la cantidad de calor requerida para cambiar la temperatura en una pequeña cantidad Δ T, o más precisamente, por una cantidad infinitesimal dT. Así, el aumento de entropía provocado por el calentamiento de una sustancia en un rango de temperaturas que no abarca una transición de fase viene dado por la suma de las cantidades C dT/T por cada incremento de temperatura dT. Esto es, por supuesto, solo la integral
\[ S_{0^o \rightarrow T^o} = \int _{o^o}^{T^o} \dfrac{C_p}{T} dt \]
Debido a que la capacidad calorífica es en sí misma ligeramente dependiente de la temperatura, las determinaciones más precisas de entropías absolutas requieren que la dependencia funcional de C sobre T se use en la integral anterior en lugar de una C constante.
\[ S_{0^o \rightarrow T^o} = \int _{o^o}^{T^o} \dfrac{C_p(T)}{T} dt \]
Cuando esto no se conoce, se puede tomar una serie de mediciones de la capacidad calorífica en incrementos estrechos de temperatura Δ T y medir el área debajo de cada sección de la curva en la Figura\(\PageIndex{3}\).
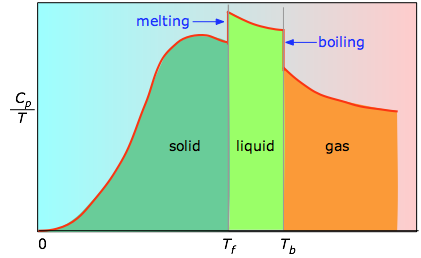
El área bajo cada sección de la gráfica representa el cambio de entropía asociado al calentamiento de la sustancia a través de un intervalo Δ T. A esto hay que añadir las entalpías de fusión, vaporización y de cualquier cambio de fase sólido-sólido. Los valores de C p para temperaturas cercanas a cero no se miden directamente, sino que pueden estimarse a partir de la teoría cuántica.
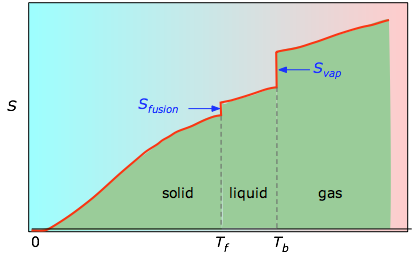
/T b se agregan para obtener la entropía absoluta a temperatura T. Como se muestra en la Figura\(\PageIndex{4}\) anterior, la entropía de una sustancia aumenta con la temperatura, y lo hace por dos razones:
- A medida que aumenta la temperatura, más microestados se vuelven accesibles, permitiendo que la energía térmica se disperse más ampliamente. Esto se refleja en el incremento gradual de la entropía con la temperatura.
- Las moléculas de sólidos, líquidos y gases tienen cada vez más libertad para moverse, facilitando la difusión y distribución de la energía térmica. Por lo tanto, los cambios de fase van acompañados de un aumento masivo y discontinuo de la entropía.
Entropias estándar de sustancias
La entropía estándar de una sustancia es su entropía a 1 atm de presión. Los valores que se encuentran en las tablas son normalmente los de 298K, y se expresan en unidades de J K —1 mol —1. La siguiente tabla muestra algunos valores típicos para sustancias gaseosas.
| Él | 126 | H 2 | 131 | CH 4 | 186 |
|---|---|---|---|---|---|
| Ne | 146 | N 2 | 192 | H 2 O (g) | 187 |
| Ar | 155 | CO | 197 | CO 2 | 213 |
| Kr | 164 | F 2 | 203 | C 2 H 6 | 229 |
| Xe | 170 | O 2 | 205 | n -C 3 H 8 | 270 |
| Cl 2 | 223 | n -C 4 H 10 | 310 |
Obsérvese especialmente cómo los valores dados en esta Tabla \(\PageIndex{2}\): ilustran estos puntos importantes:
- Si bien las energías internas estándar y entalpías de estas sustancias serían cero, las entropías no lo son. Esto se debe a que no existe una escala absoluta de energía, por lo que convencionalmente establecemos a cero las “energías de formación” de los elementos en sus estados estándar. La entropía, sin embargo, no mide la energía en sí misma, sino su dispersión entre los diversos estados cuánticos disponibles para aceptarla, y estos existen incluso en elementos puros.
- Es evidente que las entropías generalmente aumentan con el peso molecular. Para los gases nobles, esto es, por supuesto, un reflejo directo del principio de que los estados cuánticos traslacionales están más estrechamente empaquetados en moléculas más pesadas, permitiendo que sean ocupados.
- Las entropías de las moléculas diatómicas y poliatómicas muestran los efectos adicionales de los niveles cuánticos rotacionales.
| C (diamante) | C (grafito) | Fe | Pb | Na | S (rómbico) | Si | W |
|---|---|---|---|---|---|---|---|
| 2.5 | 5.7 | 27.1 | 51.0 | 64.9 | 32.0 | 18.9 | 33.5 |
Las entropías de los elementos sólidos están fuertemente influenciadas por la manera en que los átomos están unidos entre sí. El contraste entre el diamante y el grafito es particularmente llamativo; el grafito, que está formado por pilas de láminas hexagonales poco unidas, parece ser más del doble de bueno para absorber la energía térmica que el diamante, en el que los átomos de carbono están firmemente bloqueados en una celosía tridimensional, lo que permite ellos menos oportunidad de vibrar alrededor de sus posiciones de equilibrio. Al observar todos los ejemplos de la tabla anterior, notará una correlación inversa general entre la dureza de un sólido y su entropía. Así, el sodio, que se puede cortar con un cuchillo, tiene casi el doble de entropía que el hierro; la entropía mucho mayor del plomo refleja tanto su alto peso atómico como la relativa suavidad de este metal. Estas tendencias son consistentes con el principio frecuentemente expresado de que cuanto más “desordenada” una sustancia, mayor es su entropía.
| sólido | líquido | gas |
|---|---|---|
| 41 | 70 | 186 |
Los gases, que sirven como vehículos eficientes para esparcir la energía térmica en un gran volumen de espacio, tienen entrópías mucho más altas que las fases condensadas. De igual manera, los líquidos tienen entropías más altas que los sólidos debido a la multiplicidad de formas en que las moléculas pueden interactuar (es decir, almacenar energía).
Cómo depende la entropía de la concentración
A medida que una sustancia se dispersa más en el espacio, la energía térmica que transporta también se extiende sobre un volumen mayor, lo que lleva a un aumento de su entropía. Debido a que la entropía, como la energía, es una propiedad extensa, una solución diluida de una sustancia dada bien puede poseer una entropía menor que el mismo volumen de una solución más concentrada, pero la entropía por mol de soluto (la entropía molar) por supuesto siempre aumentará a medida que la solución se vuelve más diluida.
Para las sustancias gaseosas, el volumen y la presión son respectivamente medidas directas e inversas de concentración. Para un gas ideal que se expande a una temperatura constante (es decir, que absorbe calor del entorno para compensar el trabajo que realiza durante la expansión), el aumento de la entropía viene dado por
\[ \Delta S = R \ln \left( \dfrac{V_2}{V_1} \right) \label{23.2.4}\]
Nota: Si se permite que el gas se enfríe durante la expansión, la relación se vuelve más complicada y mejor se discutirá en un curso más avanzado.
Debido a que la presión de un gas es inversamente proporcional a su volumen, podemos alterar fácilmente la relación anterior para expresar el cambio de entropía asociado con un cambio en la presión de un gas perfecto:
\[ \Delta S = R \ln \left( \dfrac{P_1}{P_2} \right) \label{23.2.5}\]
Expresando el cambio de entropía directamente en concentraciones, tenemos la relación similar
\[ \Delta S = R \ln \left( \dfrac{c_1}{c_2} \right) \label{23.2.6}\]
Aunque estas ecuaciones se aplican estrictamente solo a gases perfectos y no pueden usarse en absoluto para líquidos y sólidos, resulta que en una solución diluida, el soluto a menudo puede tratarse como un gas disperso en el volumen de la solución, por lo que la última ecuación puede dar un valor bastante preciso para la entropía de dilución de una solución. Veremos más adelante que esto tiene importantes consecuencias en la determinación de las concentraciones de equilibrio en una mezcla de reacción homogénea.
Cómo se almacena la energía térmica en las moléculas
La energía térmica es la porción de energía de una molécula que es proporcional a su temperatura y, por lo tanto, se relaciona con el movimiento a escala molecular. ¿Qué tipo de movimientos moleculares son posibles? Para las moléculas monatómicas, solo hay una: el movimiento real de un lugar a otro, lo que llamamos traducción. Dado que hay tres direcciones en el espacio, todas las moléculas poseen tres modos de movimiento traslacional.
Para las moléculas poliatómicas, son posibles dos tipos adicionales de movimientos. Uno de ellos es la rotación; una molécula lineal como el CO 2 en la que todos los átomos están dispuestos a lo largo del eje x puede rotar a lo largo de los ejes y y z, mientras que las moléculas que tienen menos simetría pueden rotar alrededor de los tres ejes. Así, las moléculas lineales poseen dos modos de movimiento rotacional, mientras que las no lineales tienen tres modos rotacionales. Finalmente, las moléculas que constan de dos o más átomos pueden sufrir vibraciones internas. Para las moléculas que se mueven libremente en un gas, el número de modos o patrones vibracionales depende tanto del número de átomos como de la forma de la molécula, y aumenta rápidamente a medida que la molécula se vuelve más complicada.
Las poblaciones relativas de los estados cuantificados de energía traslacional, rotacional y vibracional de una molécula diatómica típica se representan por el grosor de las líneas en este esquema (¡no a escala!) diagrama. El sombreado coloreado indica la energía térmica total disponible a una temperatura dada. Los números en la parte superior muestran separaciones de orden de magnitud entre niveles adyacentes. Es fácilmente evidente que prácticamente toda la energía térmica reside en estados de traslación.
Observe el espaciamiento muy diferente de los tres tipos de niveles de energía. Esto es extremadamente importante porque determina el número de cuantos de energía que una molécula puede aceptar y, como muestra la siguiente ilustración, el número de diferentes formas en que esta energía puede distribuirse entre las moléculas.
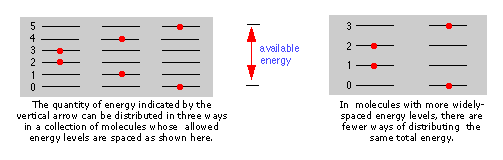
Cuanto más estrechamente estén los estados de energía cuantificados de una molécula, mayor será el número de formas en que una cantidad dada de energía térmica puede ser compartida entre una colección de estas moléculas.
El espaciamiento de los estados de energía molecular se acerca a medida que aumenta la masa y el número de enlaces en la molécula, por lo que generalmente podemos decir que cuanto más compleja es la molécula, mayor es la densidad de sus estados energéticos.
Estados cuánticos, microestados y dispersión de energía
A nivel atómico y molecular, se cuantifica toda la energía; cada partícula posee estados discretos de energía cinética y es capaz de aceptar energía térmica solo en paquetes cuyos valores corresponden a las energías de uno o más de estos estados. Las moléculas poliatómicas pueden almacenar energía en movimientos rotacionales y vibratorios, y todas las moléculas (incluso las monatómicas) poseerán energía cinética traslacional (energía térmica) a todas las temperaturas por encima del cero absoluto. La diferencia de energía entre los estados de traducción adyacentes es tan pequeña que la energía cinética traslacional puede considerarse continua (no cuantificada) para la mayoría de los propósitos prácticos.
El número de formas en que la energía térmica puede distribuirse entre los estados permitidos dentro de una colección de moléculas se calcula fácilmente a partir de estadísticas simples, pero aquí nos limitaremos a un ejemplo. Supongamos que tenemos un sistema que consta de tres moléculas y tres cuantos de energía para compartir entre ellas. Podemos darle toda la energía cinética a cualquier molécula, dejando a las otras sin ninguna, podemos dar dos unidades a una molécula y una unidad a otra, o podemos compartir la energía por igual y dar una unidad a cada molécula. Dicho todo, hay diez formas posibles de distribuir tres unidades de energía entre tres moléculas idénticas como se muestra aquí:

Cada una de estas diez posibilidades representa un microestado distinto que describirá el sistema en cualquier instante en el tiempo. Aquellos microestados que poseen distribuciones idénticas de energía entre los niveles cuánticos accesibles (y difieren solo en qué moléculas particulares ocupan los niveles) se conocen como configuraciones. Debido a que todos los microestados son igualmente probables, la probabilidad de cualquier configuración es proporcional al número de microestados que pueden producirla. Así, en el sistema mostrado anteriormente, la configuración etiquetada ii se observará 60% del tiempo, mientras que iii ocurrirá solo 10% del tiempo.
A medida que aumenta el número de moléculas y el número de cuantos, el número de microestados accesibles crece explosivamente; si 1000 cuanta de energía son compartidos por 1000 moléculas, el número de microestados disponibles será de alrededor de 10 600, un número que supera en gran medida el número de átomos en el observable universo! El número de configuraciones posibles (como se definió anteriormente) también aumenta, pero de tal manera que se reduce en gran medida la probabilidad de todas las configuraciones excepto las más probables. Así, para una muestra de un gas lo suficientemente grande como para ser observable en condiciones normales, solo se necesita considerar una única configuración (distribución de energía entre los estados cuánticos); incluso la segunda configuración más probable puede ser descuidada.
La conclusión: cualquier colección de moléculas lo suficientemente grandes en número como para tener importancia química tendrá su energía terrmal distribuida sobre un número inimaginablemente grande de microestados. El número de microestados aumenta exponencialmente a medida que más estados de energía (“configuraciones” como se definió anteriormente) se vuelven accesibles debido a
- Adición de cuantos de energía (temperatura más alta),
- Incremento en el número de moléculas (resultante de la disociación, por ejemplo).
- el volumen del sistema aumenta (lo que disminuye el espaciamiento entre los estados energéticos, permitiendo que más de ellos sean poblados a una temperatura dada.)
Muerte por calor: la difusión de energía cambia el mundo
La energía se conserva; si levantas un libro de la mesa, y lo dejas caer, la cantidad total de energía en el mundo permanece sin cambios. Todo lo que has hecho es transferirlo de la forma en que estaba almacenada dentro de la glucosa en tu cuerpo a tus músculos, y luego al libro (es decir, trabajaste en el libro moviéndolo hacia arriba contra el campo gravitacional de la tierra). Después de que el libro ha caído, esta misma cantidad de energía existe que la energía térmica (calor) en el libro y la mesa.
Lo que ha cambiado, sin embargo, es la disponibilidad de esta energía. Una vez que la energía se ha extendido a la enorme cantidad de microestados térmicos en los objetos calentados, la probabilidad de que se desdisperse espontáneamente (es decir, por casualidad) es esencialmente cero. Así, aunque la energía sigue “ahí”, está para siempre más allá de la utilización o recuperación. La profundidad de esta conclusión se reconoció alrededor de 1900, cuando se describió por primera vez en la “muerte por calor” del mundo. Esto se refiere al hecho de que cada proceso espontáneo (esencialmente cada cambio que ocurre) va acompañado de la “dilución” de la energía. La implicación obvia es que toda la energía cinética a nivel molecular se extenderá por completo, y nunca sucederá nada más.
¿Por qué los gases tienden a expandirse, pero nunca contraerse?
Todo el mundo sabe que un gas, si se deja solo, tenderá a expandirse y llenar el volumen dentro del cual está confinado completa y uniformemente. ¿Qué “impulsa” esta expansión? En el nivel más simple es claro que con más espacio disponible, los movimientos aleatorios de las moléculas individuales inevitablemente las dispersarán por todo el espacio. Pero como mencionamos anteriormente, los estados energéticos permitidos que las moléculas pueden ocupar están más espaciados en un volumen mayor que en uno más pequeño. Cuanto mayor sea el volumen disponible para el gas, mayor será el número de microestados que puede ocupar su energía térmica. Dado que todos estos estados dentro del rango de energías térmicamente accesibles son igualmente probables, la expansión del gas puede verse como consecuencia de la tendencia de la energía térmica a difundirse y compartirse lo más ampliamente posible. Una vez que esto ha sucedido, la probabilidad de que este reparto de energía se revierta (es decir, que el gas se contraiga espontáneamente) es tan mínima como para ser impensable.
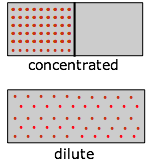
Imagínese un gas inicialmente confinado a la mitad de una caja (Figura\(\PageIndex{7}\)). Luego se retira la barrera para que pueda expandirse en todo el volumen del contenedor. Sabemos que la entropía del gas aumentará a medida que la energía térmica de sus moléculas se propague al espacio agrandado. En cuanto a la dispersión de la energía térmica, la Figura 23.2.X puede ser útil. La tendencia de un gas a expandirse se debe a los estados de energía térmica más espaciados en el mayor volumen .
.
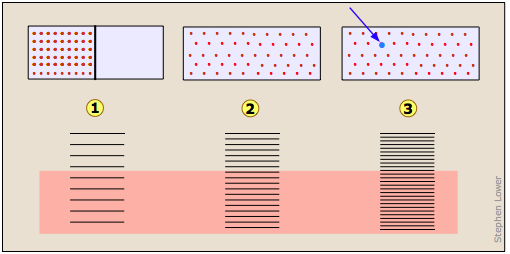
Entropía de mezcla y dilución
Mezclar y diluir realmente equivalen a lo mismo, especialmente para los gases de idea. Reemplazar el par de recipientes mostrados anteriormente por uno que contenga dos tipos de moléculas en las secciones separadas (Figura\(\PageIndex{9}\)). Cuando eliminemos la barrera, las moléculas “rojas” y “azules” se expandirán cada una en el espacio de la otra. (Recordemos la Ley de Dalton de que “cada gas es un vacío al otro gas”.) No obstante, observe que aunque cada gas sufrió una expansión, el proceso global equivale a lo que llamamos “mezcla”.
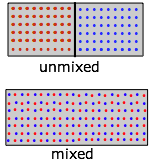
Lo que es cierto para las moléculas gaseosas puede, en principio, aplicarse también a las moléculas de soluto disueltas en un disolvente. Pero hay que tener en cuenta que mientras que la entalpía asociada a la expansión de un gas perfecto es por definición cero, Δ H de mezcla de dos líquidos o de disolución de un soluto en un disolvente tienen valores finitos que pueden limitar la miscibilidad de líquidos o la solubilidad de un soluto. Pero lo que es realmente dramático es que cuando solo se introduce una molécula de un segundo gas en el contenedor ( en la Figura\(\PageIndex{8}\)), se hace posible un número inimaginablemente enorme de nuevas configuraciones, lo que aumenta enormemente el número de microestados que son térmicamente accesibles (como se indica por el sombreado rosado de arriba).
en la Figura\(\PageIndex{8}\)), se hace posible un número inimaginablemente enorme de nuevas configuraciones, lo que aumenta enormemente el número de microestados que son térmicamente accesibles (como se indica por el sombreado rosado de arriba).
Por qué el calor fluye de caliente a frío
Así como los gases cambian espontáneamente sus volúmenes de “más pequeños a grandes”, el flujo de calor de un cuerpo más cálido a uno más frío siempre opera en la dirección de “más caliente a más frío” porque esto permite que la energía térmica pueble un mayor número de microestados de energía a medida que se ponen a disposición otros nuevos al traer el cuerpo más frío en contacto con el más cálido; en efecto, la energía térmica se vuelve más “diluida”.
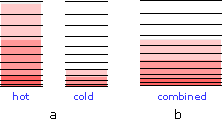
Cuando los cuerpos se ponen en contacto térmico (b), la energía térmica fluye desde los niveles más altos ocupados en el objeto más cálido hacia los niveles desocupados del uno más frío hasta que se ocupan números iguales en ambos cuerpos, llevándolos a la misma temperatura. Como cabría esperar, el aumento en la cantidad de energía que se esparce y comparte es proporcional a la cantidad de calor transferido q, pero hay otro factor involucrado, y esa es la temperatura a la que se produce la transferencia. Cuando una cantidad de calor q pasa a un sistema a temperatura T, el grado de dilución de la energía térmica viene dado por
\[\dfrac{q}{T}\]
Para entender por qué tenemos que dividir por la temperatura, consideremos el efecto de valores muy grandes y muy pequeños de T en el denominador. Si el cuerpo que recibe el calor está inicialmente a una temperatura muy baja, inicialmente se ocupan relativamente pocos estados de energía térmica, por lo que la cantidad de energía que se extiende a estados vacantes puede ser muy grande. Por el contrario, si la temperatura es inicialmente grande, ya se esparce más energía térmica en su interior, y la absorción de la energía adicional tendrá un efecto relativamente pequeño en el grado de trastorno térmico dentro del cuerpo.
Reacciones químicas: por qué la constante de equilibrio depende de la temperatura
Cuando se produce una reacción química, se involucran dos tipos de cambios relacionados con la energía térmica:
- Las formas en que se puede almacenar la energía térmica dentro de los reactivos generalmente serán diferentes de las de los productos. Por ejemplo, en la reacción H 2 → 2 H, el reactivo dihidrógeno posee estados de energía vibracional y rotacional, mientras que el hidrógeno atómico en el producto solo tiene estados de traslación, pero el número total de estados de traslación en dos moles de H es dos veces mayor que en un mol de H 2 . Debido a su espaciado extremadamente cercano, los estados de traslación son los únicos que realmente cuentan a temperaturas ordinarias, por lo que podemos decir que la energía térmica puede llegar a diluirse dos veces (“esparcirse”) en el producto que en el reactivo. Si este fuera el único factor a considerar, entonces la disociación del dihidrógeno siempre sería espontánea y esta molécula no existiría.
- Para que esta disociación ocurra, sin embargo, se debe tomar del entorno una cantidad de energía térmica (calor) q =Δ U para romper el enlace H-H. Es decir, el estado fundamental (la energía a la que comienza el colector de estados energéticos) es mayor en H, como lo indica el desplazamiento vertical de la mitad derecha en cada uno de los cuatro paneles inferiores.
En la Figura\(\PageIndex{11}\) a re representaciones esquemáticas de los niveles de energía traslacional de los dos componentes H y H2 de la reacción de disociación de hidrógeno. El sombreado muestra cómo las poblaciones relativas de microestados ocupados varían con la temperatura, provocando que la composición de equilibrio cambie a favor del producto de disociación.
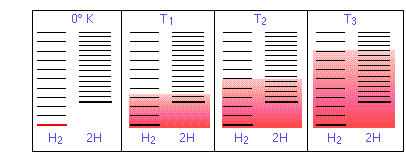
La capacidad de la energía para propagarse en las moléculas del producto está limitada por la disponibilidad de energía térmica suficiente para producir estas moléculas. Aquí es donde entra la temperatura. A cero absoluto la situación es muy simple; no hay energía térmica disponible para provocar la disociación, por lo que el único componente presente será el dihidrógeno.
- A medida que aumenta la temperatura, aumenta el número de estados energéticos poblados, como lo indica el sombreado en el diagrama. A temperatura T 1, el número de estados poblados de H 2 es mayor que el de 2H, por lo que algunos de estos últimos estarán presentes en la mezcla de equilibrio, pero sólo como componente minoritario.
- A alguna temperatura T 2 los números de estados poblados en los dos componentes del sistema de reacción serán idénticos, por lo que la mezcla de equilibrio contendrá H 2 y “2H” en cantidades iguales; es decir, la relación molar de H 2/H será 1:2.
- A medida que la temperatura sube a T 3 y superiores, vemos que el número de estados energéticos que son térmicamente accesibles en el producto comienza a superar el del reactivo, favoreciendo así la disociación.
El resultado es exactamente lo que predice el Principio LeChatelier: el estado de equilibrio para una reacción endotérmica se desplaza hacia la derecha a temperaturas más altas.
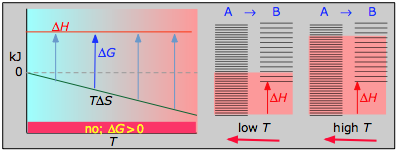
La siguiente tabla generaliza estas relaciones para las cuatro combinaciones de signos de Δ H y Δ S. (Obsérvese que el uso de los valores estándar Δ H° y Δ S° en las reacciones de ejemplo no es estrictamente correcto aquí, y puede producir resultados engañosos cuando se usan generalmente).
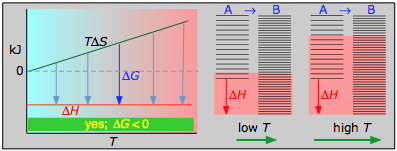
Esta reacción de combustión, como la mayoría de tales reacciones, es espontánea a todas las temperaturas. El cambio de entropía positivo se debe principalmente a la mayor masa de moléculas de CO 2 en comparación con las de O 2.
< 0
- Δ H° = —46.2 kJ
- Δ S° = —389 J K —1
- Δ G° = —16.4 kJ a 298 K
La disminución de moles de gas en la síntesis de amoníaco de Haber impulsa el cambio de entropía negativo, haciendo que la reacción sea espontánea solo a bajas temperaturas. Así, una T más alta, que acelera la reacción, también reduce su extensión.
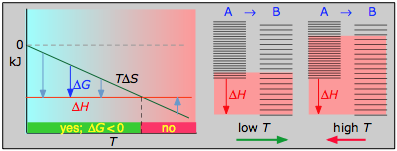
> 0

- Δ H° = 55.3 kJ
- Δ S° = +176 J K —1
- Δ G° = +2.8 kJ a 298 K
Las reacciones de disociación son típicamente endotérmicas con cambio de entropía positivo y, por lo tanto, son espontáneas a altas temperaturas. En última instancia, todas las moléculas se descomponen en sus átomos a temperaturas suficientemente altas.
< 0
- Δ H° = 33.2 kJ
- ΔS° = —249 J K — 1
- Δ G° = +51.3 kJ a 298 K
Esta reacción no es espontánea a ninguna temperatura, lo que significa que su reverso siempre es espontáneo. Pero debido a que la reacción inversa está cinéticamente inhibida, el NO 2 puede existir indefinidamente a temperaturas ordinarias aunque sea termodinámicamente inestable.
Cambios de fase
Todo el mundo sabe que el sólido es la forma estable de una sustancia a bajas temperaturas, mientras que el estado gaseoso prevalece a altas temperaturas. ¿Por qué debería ser esto? El diagrama de la Figura\(\PageIndex{12}\) muestra que
- la densidad de los estados energéticos es menor en el sólido y mayor (mucho, mucho mayor) en el gas, y
- los estados terrestres del líquido y del gas son compensados con respecto al estado anterior por los calores de fusión y vaporización, respectivamente.
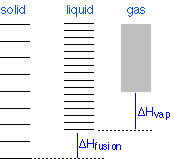
Los cambios de fase implican intercambio de energía con el entorno (cuyo contenido energético relativo al sistema se indica (¡con mucha exageración!) por la altura de las barras verticales amarillas en la Figura\(\PageIndex{13}\). Cuando el sólido y el líquido están en equilibrio (sección media del diagrama a continuación), hay suficiente energía térmica (indicada por sombreado rosado) para poblar los estados energéticos de ambas fases. Si se permite que el calor fluya hacia el entorno, se retira selectivamente de los niveles más abundantemente poblados de la fase líquida, haciendo que la cantidad de esta fase disminuya a favor del sólido. La temperatura permanece constante ya que el calor de fusión se devuelve al sistema en compensación exacta por el calor perdido en los alrededores. Por último, después de que el último rastro de líquido haya desaparecido, los únicos estados que quedan son los del sólido. Cualquier extracción adicional de calor da como resultado una caída de temperatura a medida que los estados del sólido se despoblan.

Propiedades coligativas de las soluciones
La disminución de la presión de vapor, la elevación del punto de ebullición, la depresión del punto de congelación y la ósmosis son fenómenos bien conocidos que ocurren cuando un soluto no volátil como el azúcar o una sal se disuelve en un disolvente volátil como el agua. Todos estos efectos resultan de la “dilución” del disolvente por el soluto agregado, y debido a esta comunalidad se les conoce como propiedades coligativas (Lat. co ligare, conectado a.) El papel clave de la concentración de disolvente se ve oscurecido por las expresiones muy simplificadas utilizadas para calcular la magnitud de estos efectos, en las que solo aparece la concentración de soluto. Los detalles de cómo llevar a cabo estos cálculos y las muchas aplicaciones importantes de las propiedades coligativas se cubren en otra parte. Nuestro propósito aquí es ofrecer una explicación más completa de por qué ocurren estos fenómenos.
Básicamente, todos estos resultan del efecto de dilución del disolvente sobre su entropía, y así en el incremento en la densidad de estados energéticos del sistema en la solución respecto a la del líquido puro. El equilibrio entre dos fases (líquido-gas para ebullición y sólido-líquido para congelación) ocurre cuando los estados energéticos en cada fase pueden ser poblados a densidades iguales. Las temperaturas a las que esto ocurre son representadas por el sombreado.

La dilución del solvente agrega nuevos estados de energía al líquido, pero no afecta la fase de vapor. Esto eleva la temperatura requerida para hacer accesibles números iguales de microestados en las dos fases.
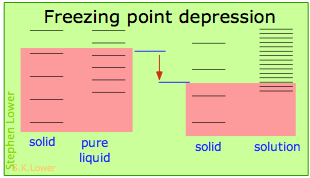
La dilución del solvente agrega nuevos estados de energía al líquido, pero no afecta a la fase sólida. Esto reduce la temperatura requerida para hacer accesible el mismo número de estados en las dos fases.
Efectos de la presión sobre la entropía: Presión osmótica
Cuando un líquido es sometido a presión hidrostática, por ejemplo, por un gas inerte y no disolvente que ocupa el espacio de vapor por encima de la superficie, se eleva la presión de vapor del líquido (Figura\(\PageIndex{16}\)). La presión actúa comprimiendo el líquido muy ligeramente, estrechando efectivamente el pozo de energía potencial en el que residen las moléculas individuales y aumentando así su tendencia a escapar de la fase líquida. (Debido a que los líquidos no son muy compresibles, el efecto es bastante pequeño; una presión aplicada de 100 atm elevará la presión de vapor del agua a 25°C en solo aproximadamente 2 torr.) En cuanto a la entropía, podemos decir que la presión aplicada reduce las dimensiones de la “caja” dentro de la cual los principales movimientos de traslación de las moléculas están confinados dentro del líquido, reduciendo así la densidad de estados energéticos en la fase líquida.
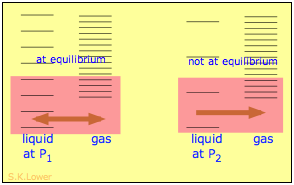
La aplicación de presión hidrostática a un líquido aumenta el espaciamiento de sus microestados, de manera que el número de estados energéticamente accesibles en el gas, aunque sin cambios, es relativamente mayor, aumentando así la tendencia de las moléculas a escapar a la fase de vapor. En términos de energía libre, la mayor presión eleva la energía libre del líquido, pero no afecta la de la fase gaseosa.

Este fenómeno puede explicar la presión osmótica. La presión osmótica, se debe recordar a los estudiantes, no es lo que impulsa la ósmosis, sino más bien la presión hidrostática que se debe aplicar a la solución más concentrada (disolvente más diluido) para detener el flujo osmótico de disolvente hacia la solución. El efecto de esta presión\(\Pi\) es aumentar ligeramente el espaciamiento de los estados de energía del disolvente en el lado de alta presión (dilución-disolvente) de la membrana para que coincida con el del disolvente puro, restaurando el equilibrio osmótico.