
15.4: Energía Libre y la Función Gibbs
- Page ID
- 71054

- Gibbs Energy es una función de estado definida como\(G = H – TS\).
- La utilidad práctica de la función Gibbs es que\(ΔG\) para cualquier proceso es negativo si conduce a un incremento en la entropía del mundo. Así, el cambio espontáneo a una temperatura y presión dadas solo puede ocurrir cuando conduciría a una disminución de\(G\).
- El signo del cambio estándar de energía libre\(ΔG^o\) de una reacción química determina si la reacción tenderá a continuar en la dirección hacia adelante o hacia atrás.
- De igual manera, los signos relativos de\(ΔH^o\) y\(ΔH^o\) determinan si la espontaneidad de una reacción química se verá afectada por la temperatura, y si es así, de qué manera.
- La existencia de puntos de fusión y ebullición agudos refleja las diferentes dependencias de temperatura de las energías libres de las fases sólida, líquida y vapor de una sustancia pura, que a su vez reflejan sus diferentes entropías.
Anteriormente, vimos que es la suma de los cambios de entropía del sistema y entorno lo que determina si un proceso ocurrirá espontáneamente. En la termodinámica química preferimos centrar nuestra atención en el sistema más que en el entorno, y nos gustaría evitar tener que calcular explícitamente el cambio de entropía del entorno.
En esta unidad se introduce una nueva función termodinámica, la energía libre, que resulta ser el criterio más útil para predecir la dirección de una reacción química y la composición del sistema en equilibrio. Sin embargo, el término “energía libre”, aunque todavía se usa ampliamente, es bastante engañoso, por lo que a menudo nos referiremos a él como “energía Gibbs”. La energía libre nos permite hacer esto por cambios que ocurren a una temperatura y presión constantes (la energía Gibbs) o temperatura y volumen constantes (la energía Helmholtz).
Energía libre: la función Gibbs
La energía de Gibbs (también conocida como la función Gibbs o Potencial de Gibbs) se define como
\[G = H – T S \label{23.4.1}\]
en el que\(S\) se refiere a la entropía del sistema. Ya que\(H\),\(T\) y\(S\) son todas las funciones estatales, así es\(G\). Así, para cualquier cambio de estado (bajo temperatura constante), podemos escribir la relación extremadamente importante
\[ΔG = ΔH – T ΔS \label{23.4.2}\]
¿Cómo engloba esta sencilla ecuación el cambio de entropía del mundo\(ΔS_{total}\), que ya sabemos que es el único criterio para el cambio espontáneo a partir de la segunda ley de la termodinámica? Empezando por la definición
\[ΔS_{total} = ΔS_{surr} + ΔS_{sys} \label{23.4.3}\]
primero nos gustaría deshacernos de\(ΔS_{surr}\). ¿Cómo puede una reacción química (un cambio en el sistema) afectar la entropía del entorno? Debido a que la mayoría de las reacciones son exotérmicas o endotérmicas, van acompañadas de un flujo de calor q p a través del límite del sistema. El cambio de entalpía de la reacción\(ΔH\) se define como el flujo de calor hacia el sistema desde el entorno cuando la reacción se lleva a cabo a presión constante, por lo que el calor retirado del entorno será el\(–q_p\) que provocará que la entropía del entorno cambie por\(–q_p / T = –ΔH/T\). Por lo tanto, podemos reescribir la ecuación\(\ref{23.4.3}\) como
\[ΔS_{total} = \dfrac{- ΔH}{T} + ΔS_{sys} \label{23.4.4}\]
Multiplicando cada lado por\(-T\), obtenemos
\[-TΔS_{total} = ΔH - TΔS_{sys} \label{23.4.5}\]
que expresa el cambio entropía del mundo en términos de propiedades termodinámicas del sistema exclusivamente. Si\(-TΔS_{total}\) se denota por\(ΔG\), entonces tenemos Ecuación\(\ref{23.4.2}\) que define el cambio de energía de Gibbs para el proceso.
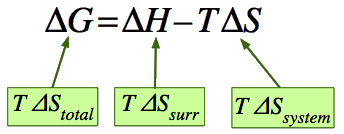
De lo anterior, debes convencerte de que\(G\) disminuirá en cualquier proceso que ocurra a temperatura y presión constantes lo cual va acompañado de un incremento general en la entropía. La temperatura constante es consecuencia de la temperatura y la entalpía que aparecen en la Ecuación anterior\(\ref{23.4.5}\). Dado que la mayoría de los cambios químicos y de fase de interés para los químicos tienen lugar bajo tales condiciones, la energía de Gibbs es la más útil de todas las propiedades termodinámicas de una sustancia, y (como veremos en la lección que sigue a esta) está estrechamente ligada a la constante de equilibrio.
Algunos libros de texto y maestros dicen que la energía libre, y por lo tanto la espontaneidad de una reacción, depende tanto de los cambios de entalpía como de entropía de una reacción, y a veces incluso se refieren a reacciones como “impulsadas por la energía” o “impulsadas por la entropía” dependiendo de si el\(TΔS\) término domina\(ΔH\) o el término. Esto es técnicamente correcto, pero engañoso porque disfraza el hecho importante de que\(ΔS_{total}\), que esta ecuación expresa de manera indirecta, es el único criterio de cambio espontáneo.
Vamos a tratar sólo con la energía Gibbs en este curso. La energía libre de Helmholtz es de interés principalmente para los ingenieros químicos (cuyos procesos a escala industrial suelen estar confinados a tanques y reactores de volumen fijo) y algunos geoquímicos cuyo interés se centra en la química que ocurre en lo profundo de la superficie terrestre.
Energía Gibbs y Cambio Químico
Recuerde que\(ΔG\) es significativo sólo para los cambios en los que la temperatura y la presión permanecen constantes. Estas son las condiciones en las que la mayoría de las reacciones se llevan a cabo en el laboratorio; el sistema suele estar abierto a la atmósfera (presión constante) y comenzamos y terminamos el proceso a temperatura ambiente (después de que cualquier calor que hayamos agregado o que sea liberado por la reacción se haya disipado). La importancia de la función Gibbs difícilmente puede ser exagerada: sirve como la única variable maestra que determina si un cambio químico dado es termodinámicamente posible. Así, si la energía libre de los reactivos es mayor que la de los productos, la entropía del mundo aumentará cuando la reacción tenga lugar tal como está escrito, y así la reacción tenderá a darse espontáneamente. Por el contrario, si la energía libre de los productos supera a la de los reactivos, entonces la reacción no se llevará a cabo en la dirección escrita, sino que tenderá a proceder en sentido inverso.
\(ΔG\)sirve como la única variable maestra que determina si un cambio químico dado es termodinámicamente posible. Además, determina la dirección y extensión del cambio químico.
En un cambio espontáneo, la energía de Gibbs siempre disminuye y nunca aumenta. Esto por supuesto refleja el hecho de que la entropía del mundo se comporta exactamente de manera opuesta (debido al signo negativo en el\(TΔS\) término).
\[\ce{H_2O(l) \rightarrow H2O(s)} \label{23.5.6}\]
el agua por debajo de su punto de congelación sufre una disminución en su entropía, pero el calor liberado al entorno lo compensa con creces, por lo que la entropía del mundo aumenta, la energía libre del H 2 O disminuye, y el proceso avanza espontáneamente.
En un cambio espontáneo, la energía de Gibbs siempre disminuye y nunca aumenta.
Una consecuencia importante de la trayectoria descendente unidireccional de la energía libre es que una vez que alcanza su valor mínimo posible, todo cambio neto se detiene. Esto, por supuesto, representa el estado de equilibrio químico. Estas relaciones se resumen muy bien de la siguiente manera:
- \(ΔG < 0\): reacción puede proceder espontáneamente a la derecha:\[A \rightarrow B \nonumber\]
- \(ΔG > 0\): reacción puede proceder espontáneamente a la izquierda:\[A \leftarrow B \nonumber\]
- \(ΔG = 0\): la reacción está en equilibrio y ambas\([A]\) y no\([B]\) va a cambiar:\[A \rightleftharpoons B. \nonumber\]
¡No es necesario encontrar el valor de ΔG para una Reacción Específica!
Esto puede parecer extraño, dada la importancia\(ΔG\) clave para determinar si una reacción tendrá lugar o no en una dirección determinada. Resulta, sin embargo, que casi nunca es necesario evaluar explícitamente\(ΔG\). Como demostraremos en la lección que sigue a esta, es mucho más conveniente trabajar con la constante de equilibrio de una reacción, dentro de la cual\(ΔG\) se “oculta”. Esto es igual de bien, porque para la mayoría de las reacciones (las que tienen lugar en soluciones o mezclas de gases) el valor de\(ΔG\) depende de las proporciones de los diversos componentes de reacción en la mezcla; no es una simple suma del tipo “productos menos reactivos”, como es el caso con\(ΔH\).
Recordando la condición para el cambio espontáneo
\[ΔG = ΔH – TΔS < 0\]
es evidente que la dependencia de la temperatura de Δ G depende casi en su totalidad del cambio de entropía asociado al proceso. (Decimos “casi” porque los valores de\(ΔH\) y\(ΔS\) son ellos mismos ligeramente dependientes de la temperatura; ambos aumentan gradualmente con la temperatura). En particular, observe que en la ecuación anterior el signo del cambio de entropía determina si la reacción se vuelve más o menos espontánea a medida que se eleva la temperatura. Para cualquier reacción dada, el signo de también\(ΔH\) puede ser positivo o negativo. Esto significa que existen cuatro posibilidades para la influencia que la temperatura puede tener en la espontaneidad de un proceso:
Caso 1: Δ H < 0 y Δ S > 0
Tanto los\(-T\Delta S\) términos entálpicos\(\Delta H\) como los entrópicos serán negativos, por lo que\(ΔG\) serán negativos independientemente de la temperatura. Una reacción exotérmica cuya entropía aumenta será espontánea a todas las temperaturas.
.svg)
Caso 2: Δ H < 0 y Δ S < 0
Si la reacción es suficientemente exotérmica puede forzar\(ΔG\) negativo solo a temperaturas por debajo de las cuales\(|TΔS| < |ΔH|\). Esto significa que hay una\(T = ΔH / ΔS\) temperatura a la que la reacción está en equilibrio; la reacción sólo procederá espontáneamente por debajo de esta temperatura. La congelación de un líquido o la condensación de un gas son los ejemplos más comunes de esta condición.
.svg)
Caso 3: Δ H > 0 y Δ S > 0
Esto es lo contrario del caso anterior; el incremento de entropía debe superar el handicap de un proceso endotérmico para que\(TΔS > ΔH\). Dado que el efecto de la temperatura es “magnificar” la influencia de un positivo\(ΔS\), el proceso será espontáneo a temperaturas superiores\(T = ΔH / ΔS\). (Piense en fundir y hervir.)

Caso 4: Δ H > 0 y Δ S < 0
Con ambos\(ΔH\) y\(ΔS\) trabajando en su contra, este tipo de procesos no procederá espontáneamente a ninguna temperatura. La sustancia A siempre tiene un mayor número de estados energéticos accesibles, y por lo tanto siempre es la forma preferida.

Las parcelas anteriores son las importantes; no trates de memorizarlas, pero asegúrate de entenderlas y de poder explicarlas o reproducirlas para un conjunto dado de Δ H y Δ S.
- Sus características diferenciadoras más importantes son la posición de la línea Δ H (por encima o por debajo de la línea es T Δ S), y la pendiente de esta última, que por supuesto depende del signo de Δ S.
- La reacción A → B ocurrirá espontáneamente solo cuando Δ G sea negativa (flechas azules apuntando hacia abajo).
- Las gráficas T Δ S no son líneas del todo rectas como se muestra aquí. Del mismo modo, las líneas que representan Δ H son aún más curvas.
Las otras dos gráficas de cada diagrama son solo para los comprometidos con la química.
- Cada par de diagramas de nivel de energía representa el espaciado relativo de los niveles de energía microscópicos en los reactivos y productos tal como se refleja en el valor de Δ S°. (Cuanto mayor es la entropía, más estrechamente espaciados están los microestados cuantificados).
- El sombreado rojo indica el rango de niveles de energía que son accesibles al sistema a cada temperatura. La dirección espontánea de la reacción siempre será en la dirección en la que el sombreado rojo se superponga al mayor número de niveles de energía, resultando en la dispersión máxima de la energía térmica.
- Obsérvese que los desplazamientos verticales corresponden a Δ H° para la reacción.
- Nunca olvides que es la capacidad de la energía térmica para propagarse a tantos de estos estados como sea posible lo que determina la tendencia del proceso a tener lugar. ¡Nada de esto es a escala, claro!
La energía estándar de Gibbs
Ya te han introducido los términos como\(ΔU^o\) y\(ΔH^o\) en los que el\(^o\) signo indica que todos los componentes (reactivos y productos) están en sus estados estándar. Este concepto de estados estándar es especialmente importante en el caso de la energía libre, así que tomemos unos momentos para revisarlo. Definiciones más exactas de los estados estándar convencionales se pueden encontrar en la mayoría de los libros de texto de química física. En campos especializados como la bioquímica y la oceanografía, pueden aplicarse definiciones alternativas. Por ejemplo, el “pH estándar” de cero (correspondiente a\([H^{+}] = 1\,M\)) no es práctico en bioquímica, por lo que comúnmente se emplea pH = 7. Para la mayoría de los propósitos prácticos, las siguientes definiciones son lo suficientemente buenas:
- gases: presión parcial de 1 atmósfera
- líquidos puros: el líquido bajo una presión total (hidrostática) de 1 atm.
- solutos: una concentración efectiva de 1 mol L —1 (1 mol dm —3). (Las concentraciones “efectivas” se acercan a las concentraciones reales ya que estas últimas se acercan a cero; para fines prácticos, estas pueden considerarse idénticas a concentraciones reales menores a aproximadamente 10 —4 molar).
- sólidos: el sólido puro bajo una presión de 1 atm
- En realidad no hay “temperatura estándar”, pero debido a que la mayoría de las tablas de termodinámica enumeran valores para 298.15 K (25° C), esta temperatura suele estar implícita.
- Estas mismas definiciones se aplican a las entalpías estándar y a las energías internas.
- No confunda estos estados estándar termodinámicos con la “temperatura y presión estándar” (STP) ampliamente empleada en los cálculos de la ley de gases.
Para hacer uso de las energías de Gibbs para predecir los cambios químicos, necesitamos conocer las energías libres de los componentes individuales de la reacción. Para ello podemos combinar la entalpía de formación estándar y la entropía estándar de una sustancia para obtener su energía de formación libre estándar
\[ΔG_f^o = ΔH_f^o – TΔS_f^o \label{23.4.7}\]
Recordemos que el símbolo° se refiere al estado estándar de una sustancia medida bajo las condiciones de 1 atm de presión o una concentración efectiva de 1 mol L —1 y una temperatura de 298 K. Luego determinar la energía Gibbs estándar de la reacción de acuerdo con
\[ ΔG^o = \sum ΔG_f^o \;(\text{products})– \sum ΔG_f^o \;(\text{reactants}) \label{24.4.8}\]
Al igual que con los calores estándar de formación, la energía libre estándar de una sustancia representa el cambio de energía libre asociado a la formación de la sustancia a partir de los elementos en sus formas más estables ya que existen bajo las condiciones estándar de 1 atm de presión y 298 K. Standard Gibbs energías libres de formación normalmente se encuentran directamente de tablas. Una vez que se conocen los valores para todos los reactivos y productos, el cambio de energía estándar de Gibbs para la reacción se encuentra por Ecuación\(\ref{23.4.7}\). La mayoría de las tablas de valores termodinámicos enumeran\(ΔG_f^o\) valores para sustancias comunes (por ejemplo, Tabla T2), que por supuesto siempre se pueden encontrar a partir de valores de Δ H f° y Δ S f°.
Encuentre el cambio de energía estándar de Gibbs para la reacción
\[\ce{CaCO3(s) \rightarrow CaO (s) + CO2(g)} \nonumber\]
Los\(ΔG_f^o°\) valores para los tres componentes de este sistema de reacción son\(\ce{CaCO3(s)}\): —1128 kJ mol —1, CaO (s): —603.5 kJ mol —1, CO 2 (g): —137.2 kJ mol —1.
Solución
Sustituyendo en Ecuación\(\ref{23.4.7}\), tenemos
\[ΔG^o = (–603.5 –137.2) – (–1128) kJ\, mol^{–1} = +130.9\, kJ\, mol^{–1} \nonumber \]
Esto indica que el proceso no es espontáneo en condiciones estándar (es decir, la carbona cálcica sólida no formará óxido de calcio sólido y CO 2 a una presión parcial de 1 atm a 25° C).
Comentario: Esta reacción se lleva a cabo a gran escala para fabricar cemento, por lo que es obvio que el proceso puede ser espontáneo bajo diferentes condiciones.
La importancia práctica de la energía de Gibbs es que nos permite hacer predicciones basadas en las propiedades (valores Δ G°) de los reactivos y productos mismos, eliminando la necesidad de experimentar. Pero hay que tener en cuenta que si bien la termodinámica siempre predice correctamente si un proceso dado puede tener lugar (es espontáneo en el sentido termodinámico), es incapaz de decirnos si se llevará a cabo a un ritmo observable.
Cuando la termodinámica dice “no”, significa exactamente eso. Cuando dice “sí”, significa “tal vez”.
La reacción
\[\ce{ 1/2 O2(g) + H2(g) → H2O(l)} \nonumber\]
se utiliza en celdas de combustible para producir una corriente eléctrica. La reacción también se puede llevar a cabo por combustión directa.
Datos termodinámicos: entropías molares en J mol —1 K —1: O 2 (g) 205.0; H 2 (g) 130.6; H 2 O (l) 70.0; H 2 O (l) Δ H° f = —285.9 kJ mol —1.
Utilice esta información para encontrar
- La cantidad de calor liberado cuando la reacción tiene lugar por combustión directa;
- La cantidad de trabajo eléctrico que la misma reacción puede realizar cuando se lleva a cabo en una pila de combustible a 298 K en condiciones reversibles;
- La cantidad de calor liberado en las mismas condiciones.
Solución
Primero, necesitamos encontrar\(ΔH^o\) y\(ΔS^o\) para el proceso. Recordando que la entalpía estándar de formación de los elementos es cero,
\[\begin{align*} ΔH^o &= ΔH^p_f(\text{products}) – ΔH^°_f(\text{reactants}) \\[4pt] &= –285.9\, kJ\, mol^{–1} – 0 \\[4pt] &= –285.9 \,kJ \,mol^{–1} \end{align*}.\]
Del mismo modo,
\[\begin{align*} ΔS^o &= S^o_f(\text{products}) – S^o_f(\text{reactants}) \\[4pt] &= (70.0) – (½ \times 205.0 + 130.6) \\[4pt] &= –163\, J\, K^{–1}mol^{–1} \end{align*}\]
- Cuando el hidrógeno y el oxígeno se combinan directamente, el calor liberado será\(ΔH^o = –285.9\, kJ\, mol^{–1}\).
- El trabajo eléctrico máximo que puede realizar la pila de combustible viene dado por\[\begin{align*}ΔG^o &= ΔH^o – TΔS^o \\[4pt] &= –285.9 \,kJ\, mol^{–1} – (298\, K)(–163\, JK^{–1}mol^{–1}) \\[4pt] &= –237.2 \,kJ\, mol^{–1}.\end{align*}.\]
- El calor liberado en la reacción de la pila de combustible es la diferencia entre el cambio de entalpía (la energía total disponible) y el trabajo reversible que se gastó:\[\begin{align*} ΔH^o – ΔG^o &= TΔS^o \\[4pt] &= (298\, K)(–163\, JK^{–1}mol^{–1}) \\[4pt] &= –48,800\, J\, mol^{–1} \\[4pt] &=–48.8 \,kJ\, mol^{–1}.\end{align*}.\]
El ejemplo anterior ilustra una ventaja importante de las pilas de combustible. Aunque la combustión directa de un mol de gas hidrógeno produce más energía que la producida por la misma reacción neta dentro de la celda de combustible, esta última, en forma de energía eléctrica, puede ser utilizada con una eficiencia energética de casi el 100 por ciento por un motor o algún otro dispositivo eléctrico. Si la energía térmica liberada por la combustión directa fuera suministrada a un motor térmico, las consideraciones de segunda ley requerirían que al menos la mitad de esta energía se “desperdiciara” al entorno.
Δ G° se refieren a cambios químicos únicos y específicos en los que todos los componentes (reactivos y productos) se encuentran en sus estados estándar.
El\(ΔG_f^o\) de una sustancia, como\(ΔH_f^o\), se refiere a la reacción en la que esa sustancia se forma a partir de los elementos tal como existen en sus formas más estables a 1 atm de presión y (generalmente) 298 K. Ambos términos son por definición cero para los elementos en sus estados estándar. Solo hay algunos casos comunes en los que esto podría crear cierta ambigüedad:
| Forma estable | \(ΔG_f^o\)(kJ mol —1) | Forma inestable | \(ΔG_f^o\)(kJ mol —1) |
|---|---|---|---|
| \(\ce{O2(g)}\) | \ (ΔG_f^O\) (kJ mol—1) ">0 | \(\ce{O3(g)}\) | \ (ΔG_F^O\) (kJ mol—1) ">163.2 |
| \(\ce{C(graphite)}\) | \ (ΔG_F^O\) (kJ mol—1)” style="text-align:center;” class="lt-chem-3597">0 | \(\ce{C(diamond)}\) | \ (ΔG_F^O\) (kJ mol—1)” style="text-align:center;” class="lt-chem-3597">2.9 |
| \(\ce{S(rhombic)}\) | \ (ΔG_F^O\) (kJ mol—1)” style="text-align:center;” class="lt-chem-3597">0 | \(\ce{S(monoclinic)}\) | \ (ΔG_F^O\) (kJ mol—1)” style="text-align:center;” class="lt-chem-3597">0.1 |
| \(\ce{P(white)}\) | \ (ΔG_F^O\) (kJ mol—1)” style="text-align:center;” class="lt-chem-3597">0 | \(\ce{P4(g)}\) | \ (ΔG_F^O\) (kJ mol—1)” style="text-align:center;” class="lt-chem-3597">24.4 |
Los iones en solución acuosa son un caso especial; sus energías libres estándar son relativas al ion hidrógeno hidratado\(\ce{H^{+}(aq)}\) que se le asigna\(ΔG_f^o = 0\).
\(ΔG\)es muy diferente de ΔG°. La distinción se ilustra muy bien\(\PageIndex{5}\) en la Figura en la que Δ G se representa en un eje vertical para dos reacciones hipotéticas que tienen signos opuestos de Δ G°. El eje horizontal expresa esquemáticamente las concentraciones relativas de reactivos y productos en cualquier punto del proceso. Obsérvese que el origen corresponde a la composición en la que la mitad de los reactivos han sido convertidos en productos.



Tome nota cuidadosa de lo siguiente:
- para la reacción Δ G° > 0. Observe que hay un número infinito de estos valores, dependiendo del progreso de la reacción. En contraste solo hay un único valor
 de Δ G°, correspondiente a la composición en la que Δ G = 0 (
de Δ G°, correspondiente a la composición en la que Δ G = 0 ( ).
). - En este punto, se han formado algunos productos, pero la composición sigue dominada por los reactivos.
- Si comenzamos en una composición a la izquierda de, Δ G será negativa y la composición se moverá hacia la derecha. De igual manera si comenzamos con una composición a la derecha de, Δ G será positiva y la composición se moverá hacia la izquierda.
- La gráfica de la derecha es para la reacción Δ G ° < 0, para la cual Δ G ° se muestra en
 . En su punto de equilibrio
. En su punto de equilibrio , hay más productos que reactivos. Si partimos de una composición a la derecha de, la composición tenderá a moverse hacia la izquierda. Si la composición inicial está a la izquierda de, la reacción tenderá a proceder a la derecha.
, hay más productos que reactivos. Si partimos de una composición a la derecha de, la composición tenderá a moverse hacia la izquierda. Si la composición inicial está a la izquierda de, la reacción tenderá a proceder a la derecha. - ¿Qué pasaría si Δ G° fuera 0? El punto de equilibrio de tal reacción estaría en el origen, correspondiendo a que la mitad de los reactivos se conviertan en productos.
El principio importante que debes entender a partir de esto es que un Δ G° negativo no significa que los reactivos se transformarán completamente en productos. De la misma manera, un Δ G° positivo no significa que no se formen productos en absoluto.
Ahora debería quedar claro a partir de la discusión anterior que una reacción dada llevada a cabo en condiciones estándar se caracteriza por un único valor de Δ G°.
La razón del mínimo de energía de Gibbs en equilibrio se relaciona con el aumento de la entropía cuando los productos y reactivos coexisten en la misma fase. Como se ve en la parcela, incluso una cantidad mínima de “contaminación” de los productos por los reactivos reduce la energía libre por debajo de la de los productos puros. En contraste, la composición de un sistema de reacción química experimenta un cambio continuo hasta alcanzar el estado de equilibrio. Así que la una sola reacción puede tener un número infinito de valores Δ G, reflejando las infinitas composiciones posibles entre los extremos de los reactivos puros (grado cero de reacción) y los productos puros (extensión de la unidad de reacción).
En el ejemplo de una reacción A → B, representada en el diagrama anterior, la energía libre estándar de los productos es menor que la de los reactivos, por lo que la reacción se llevará a cabo espontáneamente. T his no quiere decir que cada mol de A puro se convertirá en un mol de B puro. Para las reacciones en las que los productos y reactivos ocupan una sola fase (gas o solución), el significado de “espontáneo” es que la composición de equilibrio corresponderá a un grado de reacción mayor de 0.5 pero menor que la unidad.Obsérvese, sin embargo, que para Δ G° valores en exceso de aproximadamente ±50 kJ mol —1, la composición de equilibrio será despreciablemente diferente de cero o unidad de extensión de reacción. El significado físico de Δ G es que nos dice hasta qué punto ha cambiado la energía libre del sistema de G° de los reactivos puros. A medida que la reacción avanza hacia la derecha, la composición cambia y Δ G comienza a caer. Cuando la composición alcanza, Δ G alcanza su valor mínimo y una reacción adicional provocaría que suba. Pero debido a que la energía libre sólo puede disminuir pero nunca aumentar, esto no sucede. La composición del sistema permanece permanentemente en su valor de equilibrio.
Un diagrama G vs. extensión de reacción para una reacción no espontánea puede interpretarse de manera similar; la composición de equilibrio corresponderá a un grado de reacción mayor a cero pero menor a 0.5. En este caso, el mínimo at refleja el incremento de la entropía cuando los reactivos son “contaminados” por una pequeña cantidad de productos.
Si todo este detalle sobre Δ G parece un poco abrumador, no te preocupes: ¡todo se esconde en la constante de equilibrio y el cociente de reacción que discutimos en la siguiente lección!
Interpretación de Standard Gibbs energy chan ges
Aunque es\(ΔG\) más\(ΔG^o\) que eso sirve como criterio para el cambio espontáneo a temperatura y presión constantes,\(ΔG^o\) los valores están tan fácilmente disponibles que a menudo se utilizan para tener una idea aproximada de si un cambio químico dado es posible. Esto es práctico de hacer en algunos casos, pero no en otros:
Por lo general, funciona para reacciones como
\[\ce{4 NH_3(g) + 5 O_2(g) → 4 NO(g) + 6 H_2O(g)} \nonumber\]
con\(ΔG^o = –1,010\, kJ\).
(industrialmente importante para la fabricación de ácido nítrico) porque\(ΔG^o\) es tan negativa que la reacción será espontánea y prácticamente completa bajo casi cualquier conjunto razonable de condiciones.
La siguiente reacción expresa el hecho de que la molécula de agua es termodinámicamente estable:
\[\ce{2 H_2(g) + 1/2 O_2(g)→ H_2O(l)} \nonumber\]
con\(ΔG^o = –237.2 \,kJ\).
Tenga en cuenta que esto se refiere al agua líquida (el estado estándar de H 2 O a 25°). Si lo piensas, un estándar negativo de energía de formación de Gibbs (de la cual este es un ejemplo) puede de hecho considerarse una definición de estabilidad molecular.
De manera similar, la disociación del dihidrógeno en sus átomos es altamente improbable en condiciones estándar:
\[\ce{H_2O(g) → 2 H(g) + O(g)} \nonumber\]
con\(ΔG^o = +406.6\, kJ\).
Nuevamente, una situación análoga se aplicaría a cualquier molécula estable.
Ahora considere la disociación del tetroxido de dinitrógeno
\[\ce{N_2O_4(g) → 2 NO_2(g)} \nonumber\]
con\(ΔG^o = +2.8 kJ\).
en el que el valor positivo de Δ G° nos dice que N 2 O 4 a 1 atm de presión no cambiará a dos moles de NO 2 a la misma presión, pero debido al pequeño valor absoluto de Δ G°, podemos esperar la espontaneidad de la proceso para ser bastante sensible tanto a la temperatura (como se muestra en la tabla siguiente) como a la presión exactamente de la manera que predice el principio de Le Chatelier.
Para las reacciones que involucran iones disueltos, hay que tener bastante cuidado. Así, para la disociación del ácido fluorhídrico débil
\[\ce{HF(aq) → H^+(aq) + F^–(aq)} \nonumber\]
con\(ΔG^o = –317 \,kJ\).
está claro que una solución de HF de 1 mol/L no se disociará en iones 1M, pero este hecho no es muy útil porque si el HF se agrega al agua, la concentración inicial del ion fluoruro será cero (y la de H + muy cercana a cero), y el principio de Le Chatelier nuevamente predice que alguna disociación será espontánea.
Es de conocimiento común que la disociación del agua en iones hidrógeno e hidroxilo ocurre solo con mucha moderación:
\[\ce{H_2O(l) → H^+(aq) + OH^–(aq) } \nonumber\]
con\(ΔG^o = 79.9 \,kJ\).
que predice correctamente que el agua no formará 1M (concentración efectiva) de los iones, pero esto no es noticia si ya se sabe que el producto de estas concentraciones de iones nunca podrá superar 10 —14 a 298K.
Por último, considere este proceso más familiar de todos los procesos de cambio de fase, la vaporización del agua líquida:
\[\ce{H_2O(l) → H_2O(g)} \nonumber\]
con\(ΔG^o = 8.58 \,kJ \).
La conversión de agua líquida a su vapor a 1 atm de presión parcial no se realiza a 25° C, a cuya temperatura la presión parcial de equilibrio del vapor (la “presión de vapor”) es de solo 0.031 atm (23.8 torr.) El H 2 O gaseoso a una presión de 1 atm solo puede existir a 100 °C. Por supuesto, el agua que queda en un recipiente abierto a temperatura ambiente se evaporará espontáneamente si la presión parcial del vapor de agua en el aire es inferior a 0.031 atm, correspondiente a una humedad relativa inferior al 100%
Encontrar la temperatura de equilibrio
Una reacción está en su estado de equilibrio cuando
\[ΔG = ΔH – TΔS = 0 \label{23.4.1a}\]
La temperatura a la que esto ocurre viene dada por
\[T = \dfrac{ΔH}{TΔS} \label{23.4.1b}\]
Si aproximamos\(ΔH\) por\(ΔH^o\) y\(ΔS\) por\(ΔS^o\), entonces la Ecuación\ ref {23.4.1a} sería
\[ΔG \approx ΔH^o – TΔS^o = 0 \label{23.4.1aa}\]
Entonces podemos estimar el punto de ebullición normal de un líquido. De los siguientes datos termodinámicos para el agua:
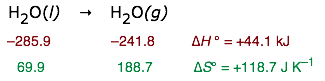
Debido a que los valores ΔH° se expresan normalmente en kilojulios mientras que ΔS° se da en julios, un error estudiantil muy común es pasar por alto la necesidad de expresar ambos en las mismas unidades.
Encontramos que el agua líquida está en equilibrio con el vapor de agua a una presión parcial de 1 atm cuando la temperatura es
\[T = \dfrac{44,100\, J}{118.7\, J\, K^{–1}} = 371.5\, K\]
Pero "el punto normal de ebullición del agua es de 373 K “, dices? Muy cierto. La razón por la que estamos fuera de aquí es que tanto Δ H° como Δ S° tienen sus propias dependencias de temperatura; estamos usando los valores “estándar” de 25° sin corregirlos a 100° C. Sin embargo, si lo piensas bien, el hecho de que podemos estimar el punto de ebullición de un líquido a partir de una tabla de ¡Los datos termodinámicos deberían ser bastante impresionantes! Por supuesto, cuanto más se acerque uno de 298 K, más poco confiable será el resultado. Así, para la disociación del dihidrógeno en sus átomos,

Todo lo que se puede decir aquí es que H 2 se descompondrá a algo más de 3000 K más o menos. (Puede que ya sepas que todas las moléculas se disociarán en sus átomos a altas temperaturas). Tendemos a pensar que las altas temperaturas de alguna manera “obligan” a las moléculas a disociarse en sus átomos, pero esto está mal. Para lograr que el enlace H-H vibre tan violentamente a través de una excitación puramente térmica que los átomos se separarían, se requeriría una temperatura más como 30,000 K. La interpretación adecuada es a la temperatura correspondiente a Δ H /TΔ S, la molécula absorbe espontáneamente energía del entorno suficiente para superar la fuerza de enlace H-H.
Predecir los efectos de la temperatura
El\(T\Delta S\) término interactúa con el\(ΔH\) término in\(\Delta G\) para determinar si la reacción puede tener lugar a una temperatura dada. Esto se puede entender más claramente examinando gráficas de\(TΔS^o\) y\(ΔH^o\) como funciones de la temperatura para algunas reacciones reales. Por supuesto, estos parámetros se refieren a estados estándar que generalmente no corresponden a las temperaturas, presiones o concentraciones que podrían ser de interés en un caso real. Sin embargo, estas cantidades se encuentran fácilmente y pueden predecir útilmente la forma en que la temperatura afecta a estos sistemas.
Caso 1: Reacción exotérmica, Δ S° > 0
\[\ce{C(graphite) + O_2(g) → CO_2(g)} \nonumber \]
- Δ H° = —393 kJ
- Δ G° = —394 kJ a 298 K

Esta reacción de combustión, como la mayoría de tales reacciones, es espontánea a todas las temperaturas. El cambio de entropía positivo se debe principalmente a la mayor masa de moléculas de CO 2 en comparación con las de O 2.
Caso 2: Reacción exotérmica, Δ S° < 0
\[\ce{3 H_2 + N_2 → 2 NH_3(g) } \nonumber\]
- Δ H° = —46.2 kJ
- Δ G° = —16.4 kJ a 298 K

La disminución de moles de gas en la síntesis de amoníaco de Haber impulsa el cambio de entropía negativo, haciendo que la reacción sea espontánea solo a bajas temperaturas. Así, una T más alta, que acelera la reacción, también reduce su extensión.
Caso 3: Reacción endotérmica, Δ S° > 0
\[ \ce{N_2O_4(g) → 2 NO_2(g)} \nonumber\]
- Δ H° = 55.3 kJ
- Δ G° = +2.8 kJ a 298 K

Las reacciones de disociación son típicamente endotérmicas con cambio de entropía positivo y, por lo tanto, son espontáneas a altas temperaturas. En última instancia, todas las moléculas se descomponen en sus átomos a temperaturas suficientemente altas.
Caso 4: Reacción Endotérmica, ΔS° < 0
\[\ce{ 1/2 N_2 (g) + O_2 (g)→ NO_2(g)} \nonumber\]
- Δ H° = 33.2 kJ
- Δ S° = —249 J K — 1
- Δ G° = +51.3 kJ a 298 K

Esta reacción no es espontánea a ninguna temperatura, lo que significa que su reverso siempre es espontáneo. Pero debido a que la reacción inversa está cinéticamente inhibida, el NO 2 puede existir indefinidamente a temperaturas ordinarias aunque sea termodinámicamente inestable.
Observaciones finales sobre Gibbs Energy
La denominación “energía libre” para G ha llevado a tanta confusión que muchos científicos ahora se refieren a ella simplemente como la energía Gibbs. La parte “libre” del nombre más antiguo refleja los orígenes del motor de vapor de la termodinámica con su interés en convertir el calor en trabajo:\(ΔG\) es la cantidad máxima de energía, que puede ser “liberada” del sistema para realizar trabajos útiles. Por “útil”, nos referimos a trabajo distinto al que está asociado con la expansión del sistema. Esto es más comúnmente en forma de trabajo eléctrico (mover la carga eléctrica a través de una diferencia de potencial), pero también son posibles otras formas de trabajo (trabajo osmótico, aumento de superficie).
Una dificultad mucho más grave con la función de Gibbs, particularmente en el contexto de la química, es que aunque G tiene las unidades de energía (julios, o en su forma intensiva, J mol —1), carece de uno de los atributos más importantes de la energía en que no es conservados. Así, aunque la energía libre siempre cae cuando un gas se expande o se produce una reacción química de manera espontánea, no es necesario que haya un aumento compensador de energía en ningún otro lugar. Referirse a G como una energía también refuerza la falsa pero generalizada noción de que una caída en la energía debe acompañar a cualquier cambio. Pero si aceptamos que la energía se conserva, es evidente que la única condición necesaria para el cambio (ya sea la caída de un peso, la expansión de un gas, o una reacción química) es la redistribución de energía. La cantidad —Δ G asociada a un proceso representa la cantidad de energía que se “comparte y difunde”, que como ya hemos explicado es el significado del incremento de la entropía. El cociente —Δ G /T es de hecho idéntico al total Δ S, el cambio de entropía del mundo, cuyo incremento es el criterio primario para cualquier tipo de cambio.
G difiere de las cantidades termodinámicas H y S de otra manera significativa: no tiene la realidad física como propiedad de la materia, mientras que H y S pueden relacionarse con la cantidad y distribución de energía en una colección de moléculas. La energía libre es simplemente una construcción útil que sirve como criterio de cambio y facilita los cálculos.
- Gibbs Energy no es energía libre
- Gibbs Energy no es energía
- Gibbs Energy ni siquiera es “real”