
15.6: Energía Libre y Equilibrio
- Page ID
- 71038

Se espera que sea capaz de definir y explicar la significación de los términos identificados en cursiva.
- A medida que avanza una reacción química homogénea, las energías Gibbs de los reactivos se vuelven más negativas y las de los productos más positivas a medida que cambia la composición del sistema.
- La energía total de Gibbs del sistema (reactantes + productos) siempre se vuelve más negativa a medida que avanza la reacción. Eventualmente alcanza un valor mínimo en una composición del sistema que define la composición de equilibrio del sistema, después de lo cual no se producirá ningún cambio neto adicional.
- La constante de equilibrio para la reacción se determina el cambio de energía estándar de Gibbs:
ΔG° = - RT ln K p - El signo de la dependencia de la temperatura de la constante de equilibrio se rige por el signo de Δ H°. Esta es la base del Principio Le Chatelier.
- Las energías Gibbs de los componentes sólidos y líquidos son constantes que no cambian con la composición. Así, en reacciones heterogéneas como los cambios de fase, la energía total de Gibbs no pasa por un mínimo y cuando el sistema no está en equilibrio solo todos los productos o todos los reactivos serán estables.
- Dos reacciones se acoplan cuando el producto de una reacción se consume en la otra. Si Δ G° para la primera reacción es positivo, el proceso general aún puede ser espontáneo si Δ G° para la segunda reacción es suficientemente negativo, en cuyo caso se dice que la segunda reacción “impulsa” la primera reacción.
Bajo condiciones de temperatura y presión constantes, el cambio químico tenderá a ocurrir en cualquier dirección que conduzca a una disminución en el valor de la energía de Gibbs. En esta lección veremos cómo G varía con la composición del sistema a medida que los reactivos se transforman en productos. Cuando G cae lo más lejos que puede, todo cambio neto llega a un alto. La composición de equilibrio de la mezcla está determinada por ΔG° que también define la constante de equilibrio K.
El camino hacia el equilibrio está por la colina de la energía de Gibbs
Esto significa, por supuesto, que si la energía total de Gibbs\(G\) de una mezcla de reactivos y productos pasa por un valor mínimo a medida que cambia la composición, entonces todo cambio neto cesará, el sistema de reacción estará en un estado de equilibrio químico. Recordará que las concentraciones relativas de reactivos y productos en el estado de equilibrio se expresan por la constante de equilibrio. En esta lección examinaremos la relación entre el cambio energético de Gibbs para una reacción y la constante de equilibrio.
Para mantener las cosas lo más simples posible, consideraremos una reacción química homogénea de la forma
\[A + B \rightleftharpoons C + D\]
en el que todos los componentes son gases a la temperatura de interés. Si la suma de las energías estándar de Gibbs de los productos es menor que la de los reactivos, Δ G° para la reacción será negativa y la reacción procederá a la derecha. Pero, ¿hasta dónde? Si los reactivos se transforman completamente en productos, la constante de equilibrio sería infinito. Las constantes de equilibrio que en realidad observamos todas tienen valores finitos, lo que implica que aunque los productos tengan una energía Gibbs menor que los reactivos, algunos de estos últimos siempre permanecerán cuando el proceso llegue al equilibrio.
Una reacción homogénea es aquella en la que todo tiene lugar en una sola fase gaseosa o líquida.
Para entender cómo las constantes de equilibrio se relacionan con los valores Δ G°, supongamos que todos los reactivos son gases, de manera que la energía Gibbs del gas A, por ejemplo, está dada en todo momento por
\[G_A = G_A^° + RT \ln P_A \label{5-1}\]
El cambio de energía de Gibbs para la reacción es la suma de las energías Gibbs de los productos, menos la suma de las energías Gibbs de los reactivos:
\[\Delta G = \underbrace{G_C + G_D}_{\text{products}} \underbrace{– G_A – G_B}_{\text{reactants}} \label{5-2}\]
Usando Ecuación\(\ref{5-1}\) para expandir cada término a la derecha de Ecuación\ ref {5-2}, tenemos
\[\Delta G = (G^°_C + RT \ln P_C) + (G^°_D + RT \ln P_D) – (G^°_B + RT \ln P_B) – (G^°_A + RT \ln P+A) \label{5-3}\]
Ahora podemos expresar los\(G^°\) términos colectivamente como\(\Delta G^°\), y combinar los términos de presión logarítmica en una sola fracción
\[ \Delta G = \Delta G° + RT \ln \left( \dfrac{P_CP_D}{P_AP_B} \right) \label{5-4}\]
que se expresa más convenientemente en términos del cociente de reacción\(Q\).
\[\Delta{G} = \Delta G^° + RT \ln Q \label{5-5}\]
La energía Gibbs\(G\) es una cantidad que se vuelve más negativa durante el transcurso de cualquier proceso natural. Así como una reacción química toma plac e,\(G\) solo cae y nunca llegará a ser más positiva. Eventualmente se alcanza un punto en el que cualquier transformación adicional de los reactivos en productos provocaría\(G\) un aumento. En este punto\(G\) está en un mínimo (ver más abajo), y no se puede producir más cambio neto; la reacción está entonces en equilibrio.
Aunque las Ecuaciones\ ref {5-1} -\ ref {5-5} son estrictamente correctas solo para gases perfectos, veremos más adelante que ecuaciones de forma similar se pueden aplicar a muchas soluciones líquidas sustituyendo las concentraciones por presiones.
Considere la reacción de disociación en fase gaseosa
\[\ce{N_2O_4 \rightarrow 2 NO_2 } \nonumber\]
que es un ejemplo sencillo de las relaciones energéticas de Gibbs en una reacción homogénea.
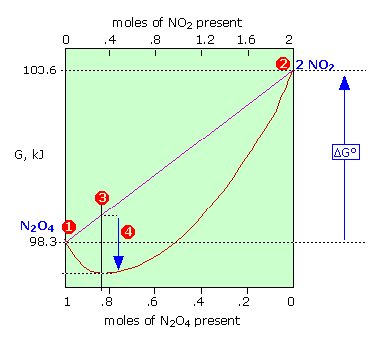
La energía Gibbs de 1 mol de N 2 O 4 (1) es menor que la de 2 moles de NO 2 (2) por 5.3 kJ; jue s\( \Delta G^o = +5.3\, \text{kJ}\) fo r la transformación completa de los reactivos en productos. La línea diagonal recta muestra la energía Gibbs de todas las composiciones posibles si se evitaba que los dos gases se mezclaran. La línea curva roja muestra la energía de Gibbs de la mezcla de reacción real. Esto pasa por un mínimo en (3) donde 0.814 mol de\(N_2O_4\) están en equilibrio con 0.372 mol de\(NO_2\). La diferencia (4) corresponde a la energía Gibbs de mezcla de reactivos y productos que siempre resulta en una mezcla de equilibrio cuya energía Gibbs es menor que la de reactivos puros o productos puros. Así ocurrirá alguna cantidad de reacción aunque Δ G° para el proceso sea positivo.
¿Cuál es la diferencia entre Δ G y Δ G°?
Es muy importante estar al tanto de esta distinción; ¡ese pequeño símbolo del° hace un mundo de diferencia! Primero, el cambio de energía estándar de Gibbs Δ G° tiene un valor único para una reacción particular a una temperatura y presión dadas; esta es la diferencia
\[ \sum G^°_{f} (\text{products}) – \sum G^°_{f}(\text{reactants}) \]
que se tabulan en tablas termodinámicas. Corresponde al cambio de energía de Gibbs para un proceso que nunca ocurre realmente: la transformación completa de N 2 O 4 puro en NO 2 puro a una presión constante de 1 atm.
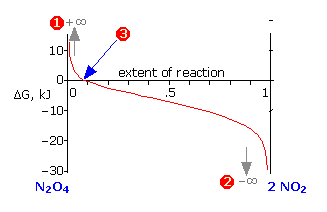
La otra cantidad\(\Delta G\), definida por la Ecuación\(\ref{5-5}\), representa las energías Gibbs totales de todas las sustancias en la mezcla de reacción en cualquier composición particular del sistema. En contraste con\(\Delta G^°\) lo que es una constante para una reacción dada,\(\Delta G\) varía continuamente a medida que cambia la composición, llegando finalmente a cero en equilibrio. \(\Delta G\)es la “distancia” (en energía de Gibbs) desde el estado de equilibrio de una reacción dada. Así para los casos limitantes de puro\(\ce{N_2O_4}\) o\(\ce{NO_2}\) (a s lejos del estado de equilibrio como puede ser el sistema!) ,
\[Q = \dfrac{[NO_2]^2}{[N_2O_4]} = \pm\infty\]
que hace que el logaritmo en la Ecuación\(\ref{5-5}\), and thus the value o f\(\Delta G\), se acerque a los mismos límites asintóticos (1) o (2). A medida que la reacción avanza en la dirección apropiada\(\Delta G\) se acerca a cero; una vez allí (3), el sistema está en su composición de equilibrio y no se producirá ningún cambio neto adicional.
El cambio de energía molar estándar de Gibbs para esta reacción muy simple es —2.26 kJ, pero la mezcla del butano sin reaccionar con el producto reduce la energía de Gibbs de la mezcla de equilibrio a aproximadamente —3.1 kJ mol —1 en la composición de equilibrio correspondiente al 77 por ciento de conversión.
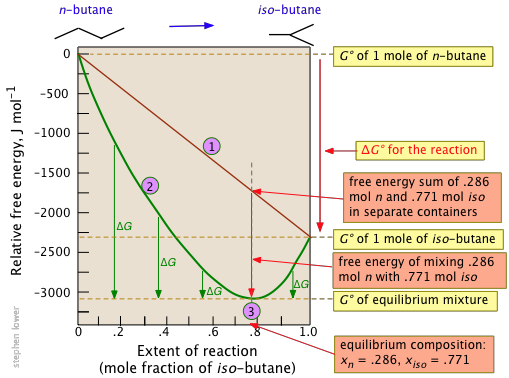
Observe particularmente que
- La suma de las energías Gibbs de los dos gases (n-butano e iso-butano) varía por separado linealmente con la composición de la mezcla (línea roja
 ).
). - La curva verde
 agrega la energía Gibbs de mezcla a la suma anterior; su mínimo define la composición de equilibrio.
agrega la energía Gibbs de mezcla a la suma anterior; su mínimo define la composición de equilibrio. - A medida que la composición se acerca al valor de equilibrio
 ,\(ΔG\) (que denota cuánto más lejos puede caer la energía Gibbs del sistema) se acerca a cero.
,\(ΔG\) (que denota cuánto más lejos puede caer la energía Gibbs del sistema) se acerca a cero.
Los cálculos detallados que conducen a los valores mostrados anteriormente se pueden encontrar aquí.
Por qué las reacciones conducen a mezclas de reactivos y productos
Ahora estamos en condiciones de responder a la pregunta planteada anteriormente: si Δ G° para una reacción es negativa, lo que significa que las energías Gibbs de los productos son más negativas que las de los reactivos, ¿por qué quedarán algunas de estas últimas después de alcanzar el equilibrio? La respuesta es que no importa cuán baja sea la energía Gibbs de los productos, la energía Gibbs del sistema se puede reducir aún más al permitir que algunos de los productos sean “contaminados” (es decir, diluidos) por algunos reactivos. Debido a la entropía asociada con la mezcla de reactivos y productos, ninguna reacción homogénea estará 100% completa. Un corolario interesante de esto es que cualquier reacción para la que se pueda escribir una ecuación química equilibrada puede, en principio, tener lugar en cierta medida, por muy minuta que sea.
Las energías Gibbs de mezcla de productos con reactivos tienden a ser bastante pequeñas, por lo que para reacciones que tienen valores Δ G° que son altamente negativos o positivos (±20 kJ mol —1, digamos), la mezcla de equilibrio será, para todos los fines prácticos, o bien reactantes [casi] “puros” o productos.
La Constante de Equilibrio
Ahora volvamos a la Ecuación\(\ref{5-5}\) que reproducimos aquí:
\[\Delta{G} = \Delta{G^°} + RT \ln Q \]
A medida que la reacción se acerca al equilibrio,\(\Delta G\) se vuelve menos negativa y finalmente llega a cero. En equilibrio\(\Delta{G} = 0\) y\(Q = K\), así podemos escribir (¡hay que saber esto!)
\[\Delta{G^°} = –RT \ln K_p \label{5-6}\]
en el que\(K_p\), la constante de equilibrio expresada en unidades de presión, es el valor especial de\(Q\) que corresponde a la composición de equilibrio.
Esta ecuación es una de las más importantes en química porque relaciona la composición de equilibrio de un sistema de reacción química con propiedades físicas medibles de los reactivos y productos. Si conoces las entropías y las entalpías de formación de un conjunto de sustancias, puedes predecir la constante de equilibrio de cualquier reacción que involucre a estas sustancias sin necesidad de saber nada sobre el mecanismo de la reacción.
En lugar de escribir Ecuación\(\ref{5-6}\) en términos de K p, podemos usar cualquiera de las otras formas de la constante de equilibrio como K c (concentraciones), K x (fracciones molares), K n (números de moles), etc. Recuerde, sin embargo, que para las soluciones iónicas especialmente, solo la K a, en la que se utilizan actividades, será estrictamente válida.
A menudo es útil resolver Ecuación\(\ref{5-6}\) para la constante de equilibrio, rindiendo
\[ K = \exp {\left ( {-\Delta G \over RT} \right )} \label{5-7}\]
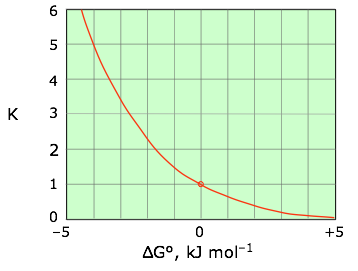

Esta relación se grafica más convenientemente contra el logaritmo de\(K\) como se muestra en la Figura\(\PageIndex{3}\), donde se puede representar como una línea recta que pasa por el punto (0,0).
Calcular la constante de equilibrio para la reacción a partir de los siguientes datos termodinámicos:
\[\ce{H^{+}(aq) + OH^{–}(aq) <=> H_2O(l)} \nonumber\]
|
\(H^+(aq)\)
|
\(OH^–(aq)\)
|
\(H_2O(l)\)
|
|
|---|---|---|---|
| ΔH f°, kJ mol —1 | \ (H^+ (aq)\) ">
0
|
\ (OH^— (aq)\) ">
—230.0
|
\ (H_2O (l)\) ">
—285.8
|
| S°, J K —1 mol —1 | \ (H^+ (aq)\) ">
0*
|
\ (OH^— (aq)\) ">
—10.9
|
\ (H_2O (l)\) ">
70.0
|
* Tenga en cuenta que la entropía estándar del ión hidrógeno es cero por definición. Esto refleja el hecho de que es imposible realizar estudios termodinámicos sobre una sola especie cargada. Todas las entropías iónicas son relativas a la de\(\ce{H^{+}(aq)}\), lo que explica por qué algunos valores (como para el ion hidróxido acuoso) son negativos.
Solución
A partir de los datos anteriores, podemos evaluar las siguientes cantidades:
\[\begin{align*} \Delta{H}^o &= \sum \Delta H^o_{f}(\text{products}) - \sum \Delta H^o_{f}(\text{reactants}) \\[4pt] &= (–285.8) - (-230) \\[4pt] &= –55.8\, kJ \; mol^{-1} \end{align*}\]
\[\begin{align*}\Delta{S}^o &= \sum \Delta S^o (\text{products}) - \sum \Delta S° (\text{reactants}) \\[4pt] &= (70.0) – (–10.9) \\[4pt] &= +80.8\, J \; K^{-1}\; mol^{-1} \end{align*}\]
El valor de\(\Delta{G}°\) a 298 K es
\[\begin{align*} \Delta H^o – T\Delta S^o &= (–55800) – (298)(80.8) \\[4pt] &= –79900\, J\, mol^{–1} \end{align*}\]
De Ecuación\(\ref{5-7}\) tenemos
\[\begin{align*} K &= \exp\left(\dfrac{–79900}{8.314 \times 298}\right) \\[4pt] &= e^{32.2} = 1.01 \times 10^{–14} \end{align*}\]
Equilibrio y Temperatura
Ya hemos discutido cómo cambiar la temperatura aumentará o disminuirá la tendencia a que se lleve a cabo un proceso, dependiendo del signo de Δ S°. Esta relación se puede desarrollar formalmente diferenciando la relación
\[ \Delta G^° = \Delta H^° – T\Delta S^° \label{5-8}\]
con respecto a la temperatura:
\[ \dfrac{d(-\Delta G^°)}{dT} = -\Delta S^° \label{5-9}\]
De ahí que el signo del cambio de entropía determine si la reacción se vuelve más o menos permitida a medida que aumenta la temperatura.
Muchas veces queremos saber cómo un cambio en la temperatura afectará el valor de una constante de equilibrio cuyo valor se conoce a alguna temperatura fija. Supongamos que la constante de equilibrio tiene el valor\(K_1\) a temperatura\(T_1\) y deseamos estimar\(K_2\) a temperatura\(T_2\). Expandiendo la ecuación\(\ref{5-7}\) en términos de\(\Delta H^°\) y\(\Delta S^°\), obtenemos
\[–RT_1 \ln K_1 = \Delta H^ ° – T_1 \Delta S^° \]
y
\[–RT_2 \ln K_2 = \Delta H ^° – T_2 \Delta S^°\]
Dividiendo ambos lados por RT y restando, obtenemos
\[ \ln K_1 - \ln K_2 = - \left( \dfrac{\Delta H^°}{RT_1} -\dfrac{\Delta H^°}{RT_2} \right) \label{5-10}\]
Que se expresa más convenientemente como la relación
\[ \ln \dfrac{K_1}{K_2} = - \dfrac{\Delta H^°}{R} \left( \dfrac{1}{T_1} -\dfrac{1}{T_2} \right) \label{5-11}\]
Esta es su base teórica del Principio de Le Ch a telier con respecto al efecto de la temperatura en el equilibrio:
- si la reacción es exotérmica\(\Delta H^° < 0\), entonces aumentar la temperatura hará que el segundo término exponencial sea más pequeño y\(K\) disminuirá. El equilibrio entonces “se desplazará hacia la izquierda”.
- Si\(\Delta H^° > 0\), entonces aumentar T hará que el exponente sea menos negativo y\(K\) aumentará y el equilibrio “se desplazará hacia la derecha”.
Esta es una relación sumamente importante, pero no sólo por su uso en el cálculo de la dependencia de temperatura de una constante de equilibrio. Aún más importante es su aplicación en la dirección “inversa” para determinar experimentalmente Δ H° a partir de dos valores de la constante de equilibrio medidos a diferentes temperaturas. Las determinaciones calorimétricas directas de los calores de reacción no son fáciles de hacer; relativamente pocos químicos tienen el equipo y la experiencia requeridos para esta tarea bastante exigente. La medición de una constante de equilibrio es generalmente mucho más fácil, y muchas veces dentro de las capacidades de cualquiera que haya tenido un curso introductorio de Química. Una vez determinado el valor de ΔH° se puede combinar con el cambio de energía de Gibbs (a partir de una sola observación de K, a través de la Ecuación\(\ref{5-7}\)) para permitir que Δ S° sea calculado a través de la Ecuación\(\ref{5-9}\).
Equilibrio sin mezclar: es todo o nada
Ahora se debe entender que para las reacciones homogéneas (aquellas que tienen lugar enteramente en fase gaseosa o en solución) la composición de equilibrio nunca será 100% productos, por más baja que sea su energía Gibbs en relación con los reactivos. Como se resumió en el ejemplo de N 2 O 4 -disociación discutido anteriormente. Esto se debe a la “dilución” de los productos por los reactivos. En reacciones heterogéneas (aquellas que involucran más de una fase) esta dilución, y los efectos que emanan de ella, pueden no ser posibles.
Un tipo de proceso heterogéneo particularmente simple pero importante es el cambio de fase. Consideremos, por ejemplo, una mezcla de equilibrio de hielo y agua líquida. La concentración de H 2 O en cada fase depende únicamente de la densidad de la fase; no hay forma de que el hielo pueda “diluirse” con agua, o viceversa. Esto significa que a todas las temperaturas que no sean el punto de congelación, el estado energético más bajo de Gibbs será el correspondiente al hielo puro o líquido puro. Sólo en el punto de congelación, donde las energías Gibbs de agua y hielo son idénticas, pueden coexistir ambas fases, y pueden hacerlo en cualquier proporción.

Energía Gibbs del sistema de hielo-agua
Solo a 0°C pueden coexistir hielo y agua líquida en cualquier proporción. Obsérvese que en contraste con el ejemplo homogéneo de N 2 O 4, no existe un mínimo de energía Gibbs en las composiciones intermedias.
Reacciones acopladas
Se dice que dos reacciones están acopladas cuando el producto de una de ellas es el reactivo en la otra:
\[A \rightarrow B \nonumber\]
y
\[B \rightarrow C \nonumber\]
Si la energía estándar de Gibbs de la primera reacción es positiva pero la de la segunda reacción es suficientemente negativa, entonces para el proceso general será negativa y decimos que la primera reacción es “impulsada” por la segunda. Esta, por supuesto, es solo otra forma de describir un efecto que ya conoces como el principio de Le Chatelier: la eliminación de la sustancia B por la segunda reacción provoca que el equilibrio de la primera se “desplace hacia la derecha”. De manera similar, la constante de equilibrio de la reacción global es el producto de las constantes de equilibrio de las dos etapas.
| 1 Cu 2 S (s) → 2 Cu (s) + S (s) | Δ G° = + 86.2 kJ | Δ H° = + 76.3 kJ |
| 2 S (s) + O 2 (g) → SO 2 (g) | Δ G° = —300.1 kJ | Δ H° = + 296.8 kJ |
| 3 Cu 2 S (s) → 2 Cu (s) + SO 2 (g) | Δ G° = —213.9 kJ | Δ H° = — 217.3 kJ |
En el ejemplo anterior, la reacción 1 es el primer paso para obtener cobre metálico a partir de uno de sus principales minerales. Esta reacción es endotérmica y tiene un cambio de energía de Gibbs positivo, por lo que no procederá espontáneamente a ninguna temperatura. Sin embargo, si el Cu 2 S se calienta en el aire, el azufre se elimina tan rápidamente como se forma por oxidación en la reacción altamente espontánea 2, que suministra la energía Gibbs requerida para impulsar 1. El proceso combinado, conocido como tostado, es de considerable importancia industrial y es uno de una gran clase de procesos empleados para obtener metales de sus minerales.