
20.14: Estructura de Proteína Primaria
- Page ID
- 75120

Las proteínas que ocurren en la naturaleza difieren entre sí principalmente porque sus cadenas laterales son diferentes. En parte se trata de una cuestión de composición. En lana, por ejemplo, 11 por ciento de las cadenas laterales son cisteína, mientras que ninguna cisteína se presenta en la seda en absoluto. Sin embargo, en un grado mucho mayor, las diferencias entre diferentes proteínas son una cuestión de la secuencia en la que ocurren las diferentes cadenas laterales. Esto es especialmente cierto de las proteínas globulares como las enzimas. Se dice que la secuencia de cadenas laterales a lo largo del esqueleto de los enlaces peptídicos en un polipéptido constituye su estructura primaria.
Los 20 aminoácidos diferentes permiten la construcción de una tremenda variedad de estructuras primarias. Consideremos, por ejemplo, cuántos tripéptidos similares a los mostrados en la Ecuación 1 de la página sobre cadenas polipeptídicas se pueden construir a partir de los 20 aminoácidos. En el ejemplo que se muestra, el primer aminoácido de la cadena es glicina, pero también podría ser prolina o cualquier otro de los 20 aminoácidos. Así, hay 20 posibilidades para el primer lugar de la cadena. De igual manera hay 20 posibilidades para el segundo lugar de la cadena, haciendo un total de 20 × 20 = 400 combinaciones posibles. Para cada una de estas 400 estructuras podemos elegir nuevamente entre 20 aminoácidos para el tercer lugar de la cadena, dando un total general de 400 × 20 = 20 3 = 8000 posibles estructuras para el tripéptido.
Una fórmula general para el número de estructuras primarias para un polipéptido que contiene n unidades de aminoácidos es 20 n, un número muy grande de hecho cuando se considera que la mayoría de las proteínas contienen al menos 50 residuos de aminoácidos. [20 50 = (2 × 10) 50 = 2 50 × 10 50 = 10 15 × 10 50 = 10 65] La estructura primaria se especifica convencionalmente escribiendo las abreviaturas de tres letras para cada aminoácido, comenzando por el extremo —NH 3 + del polímero. En algunos casos, esto incluso se acorta a la secuencia abreviada de 1 letra. Por ejemplo, la estructura
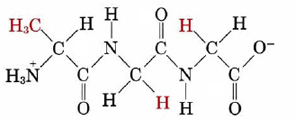
cuya lectura del extremo -NH 3 + es alanina, glicina, glicina se especificaría como
Ala-Gly-Gly o AGG
Tenga en cuenta que Ala-Gly-Gly no es lo mismo que Gly-Gly-Ala. En este último caso, la glicina más que la alanina tiene el grupo -NH 3 + libre. Debido a que sus extremos son diferentes, existe un carácter direccional en la cadena polipeptídica.
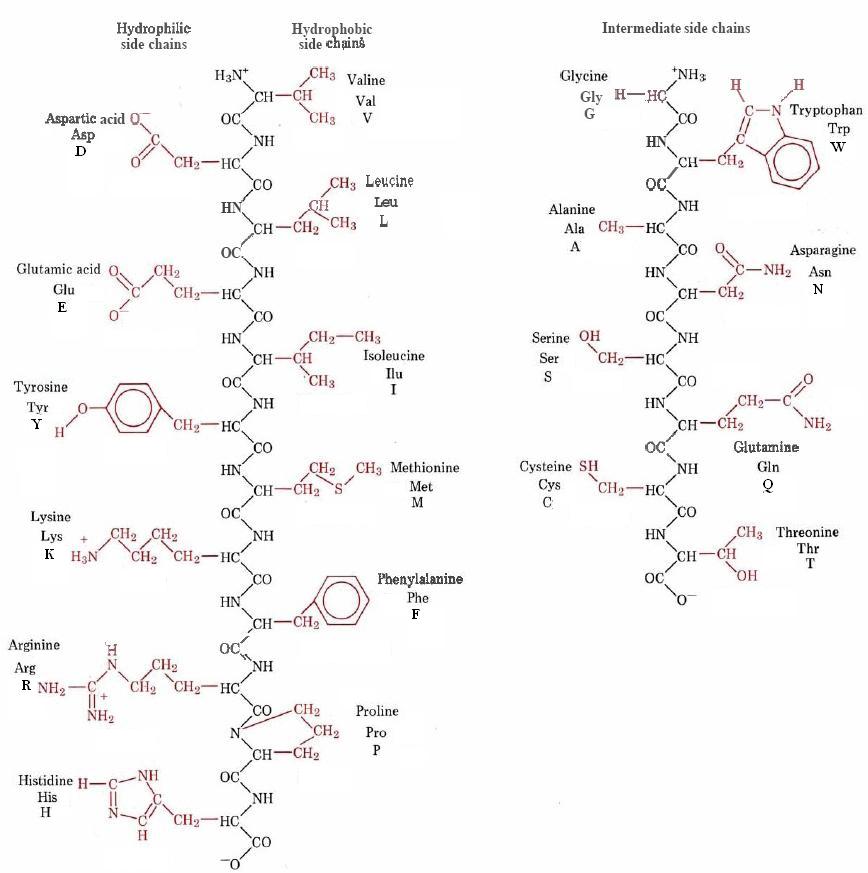
Tanto las abreviaturas de tres letras como las de una sola letra se muestran en la Figura \(\PageIndex{1}\):
La determinación de la estructura primaria de una proteína es un problema difícil y complicado. También es bastante importante: la secuencia de aminoácidos gobierna la forma tridimensional y, en última instancia, la función biológica de la proteína. En consecuencia, se ha dedicado mucho esfuerzo a los métodos mediante los cuales se puede dilucidar la estructura primaria.
La insulina fue la primera proteína cuya secuencia de aminoácidos se determinó. Esta obra pionera, concluida en 1953 tras unos 10 años de esfuerzo, le valió el Premio Nobel al bioquímico británico Frederick Sanger (nacido en 1918). Encontró que la estructura primaria era
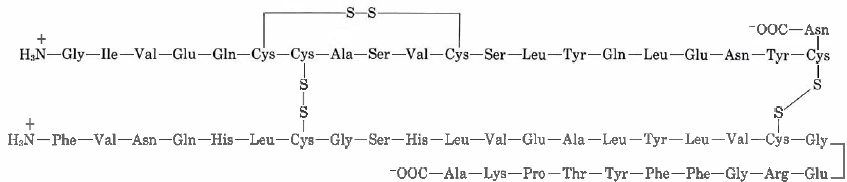
Observe cómo hay dos cadenas en esta estructura, una con 21 cadenas laterales y la otra con 30. Estas dos cadenas están unidas en dos lugares con puentes disulfuro (—S-S—), cada uno de los cuales conecta dos residuos de cisteína en diferentes cadenas.
Para determinar la estructura primaria de una proteína, primero se hierve una masa conocida de muestra pura en ácido o base hasta que se hidroliza completamente a aminoácidos individuales. La mezcla de aminoácidos se separa entonces cromatográficamente y se determina la cantidad exacta de cada aminoácido. De esta manera, se puede encontrar que por cada 3 mol de serina en la molécula de insulina, hay 6 moles de leucina. El siguiente paso es descomponer la proteína en fragmentos más pequeños. Los puentes disulfuro se rompen por oxidación después de lo cual la proteína es hidrolizada selectivamente por enzimas, llamadas proteasas, como la tripsina o la quimotripsina. En un caso favorable se tendrán entonces varios fragmentos conteniendo cada uno 10 o 20 residuos de aminoácidos. Estos se pueden separar y analizar de forma individual.
Usando la degradación de Edman, llamada así por Pehr Edman, la secuencia de aminoácidos en uno de estos fragmentos polipeptídicos generalmente se determina usando fenilsotiocianato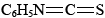 , que ataca selectivamente el extremo —NH 3 + de la cadena polipeptídica. Esta reacción se lleva a cabo en condiciones básicas. La adición de ácido luego separa el aminoácido terminal, y se puede identificar. Dado que el resto de la cadena polipeptídica se deja intacta, este proceso puede repetirse, y cada aminoácido de la secuencia puede ser atacado, eliminado e identificado. Al cortar aminoácidos uno a la vez, uno finalmente encuentra la secuencia completa para el fragmento. Todo este proceso puede automatizarse y, por lo tanto, acelerarse considerablemente. Una vez que los fragmentos han sido secuenciados, se convierte en cuestión de ordenarlos correctamente. Dado que diferentes proteasas hidrolizan enlaces peptídicos en diferentes lugares de la secuencia de aminoácidos, se pueden usar diferentes patrones de fragmentación para determinar la secuencia para la proteína completa. El extremo de un fragmento de un digesto de tripsina estará en medio de un fragmento de un digesto de quimotripsina, por ejemplo. Esto proporciona un medio relativamente rápido para secuenciar secuencias de proteínas desconocidas.
, que ataca selectivamente el extremo —NH 3 + de la cadena polipeptídica. Esta reacción se lleva a cabo en condiciones básicas. La adición de ácido luego separa el aminoácido terminal, y se puede identificar. Dado que el resto de la cadena polipeptídica se deja intacta, este proceso puede repetirse, y cada aminoácido de la secuencia puede ser atacado, eliminado e identificado. Al cortar aminoácidos uno a la vez, uno finalmente encuentra la secuencia completa para el fragmento. Todo este proceso puede automatizarse y, por lo tanto, acelerarse considerablemente. Una vez que los fragmentos han sido secuenciados, se convierte en cuestión de ordenarlos correctamente. Dado que diferentes proteasas hidrolizan enlaces peptídicos en diferentes lugares de la secuencia de aminoácidos, se pueden usar diferentes patrones de fragmentación para determinar la secuencia para la proteína completa. El extremo de un fragmento de un digesto de tripsina estará en medio de un fragmento de un digesto de quimotripsina, por ejemplo. Esto proporciona un medio relativamente rápido para secuenciar secuencias de proteínas desconocidas.
La degradación de Edman no es el único método para el cual las proteínas se secuencian hoy en día. La espectrometría de masas puede secuenciar polipéptidos de 20 a 30 aminoácidos de longitud, utilizando una técnica llamada espectrometría de masas en tándem. En este método, se envía un polipéptido a través de un espectrómetro de masas, el cual ioniza el polipéptido. El péptido cargado luego entra en una cámara de colisión, haciendo que el péptido se fragmente en diferentes enlaces peptídicos. Los fragmentos resultantes se miden luego mediante un segundo espectrómetro de masas. El espectro resultante puede determinar la secuencia peptídica por diferencias en la masa de los fragmentos.
Con el advenimiento de los métodos de secuenciación de ADN y el éxito del Proyecto Genoma Humano, muchas secuencias de proteínas ahora se determinan indirectamente, a través del código genético. Cuando se conoce la secuencia de ADN para una proteína, ésta puede ser utilizada para determinar la secuencia proteica. De la misma manera, se puede usar una secuencia proteica conocida para determinar el gen que codifica esa proteína. Por lo tanto, hay muchas formas diferentes de determinar las secuencias de proteínas, todas las cuales se complementan entre sí. [1]
A medida que los métodos para determinar la estructura primaria se han vuelto más avanzados, se han secuenciado muchas proteínas y se pueden hacer algunas comparaciones interesantes. Un ejemplo particularmente intrigante es el del citocromo c, un portador de electrones que se encuentra en todos los organismos que utilizan oxígeno para la respiración. Cuando se comparan muestras de citocromo c de diferentes organismos, se encuentra que la secuencia de aminoácidos suele ser diferente en cada caso. Además, cuanto más separadas estén las dos especies en sus características macroscópicas, mayor será el grado de diferencia en sus secuencias proteicas. Cuando se compara el citocromo c de caballo con el de la levadura, 45 de 104 residuos son diferentes. Solo se encuentran dos sustituciones entre pollo y pato, y el citocromo c es idéntico en el cerdo, la vaca y la oveja. Las magnitudes de estos cambios coinciden bastante bien con la taxonomía biológica basada en diferencias observables macroscópicamente. El citocromo c se puede utilizar para rastrear la evolución biológica desde organismos unicelulares hasta las diversas especies actuales, e incluso para estimar los tiempos en que se produjo la ramificación en el árbol genealógico de la vida. Esto hace que los métodos moleculares también sean una herramienta poderosa para el biólogo evolutivo.