
8.3: La cinética y los mecanismos de las reacciones
- Page ID
- 76516

El estudio de las velocidades de reacción, llamado cinética química, abarca una amplia gama de actividades, mediciones y cálculos. Quizás te preguntes por qué alguien se molestaría con esto, pero resulta que podemos usar datos cinéticos para obtener más información sobre una reacción que solo qué tan rápido va; podemos conocer el camino que la reacción toma de los reactivos a los productos, conocido como el mecanismo de la reacción. Si piensas en una reacción en términos moleculares, parece claro que debe haber una vía continua entre los reactivos y los productos. Los reactivos no desaparecen repentinamente y luego reaparecen como productos, y en la mayoría de las reacciones solo se rompen uno o dos enlaces y se forman a medida que avanza la reacción. Esta vía, o mecanismo, denota el orden en que se rompen y forman los enlaces, y las especies intermedias involucradas. Sin embargo, debido a que no podemos ver directamente lo que sucede a nivel molecular durante una reacción, tenemos que confiar en métodos indirectos para determinar qué está pasando. Incluso usando técnicas espectroscópicas modernas, discutidas con más detalle en la sección de espectroscopía, algunas especies en vías de reacción solo pueden estar presentes por femto (\(10^{-15}\)) o atto (\(10^{-18}\)) segundos. Los eventos en estas escalas de tiempo son difíciles de estudiar y, de hecho, gran parte de la investigación actual de vanguardia en química y física está dirigida a detectar y caracterizar tales eventos efímeros a nivel molecular. Como veremos, la información sobre cómo varía la velocidad de reacción con la concentración y la temperatura puede darnos fascinantes conocimientos químicos sobre las vías de reacción.
Concentraciones y velocidades de reacción
Como hemos visto como la probabilidad de colisiones entre moléculas reaccionantes aumenta, la velocidad de reacción aumenta. Para obtener información sobre el mecanismo de reacción necesitamos conocer la relación exacta entre concentraciones y tasas. Esto se puede hacer usando una serie de diferentes técnicas y configuraciones experimentales. Pero antes de hacer eso, tenemos que repasar algunos términos más. Recordemos que la velocidad de la reacción es el cambio en la concentración de reactivo por unidad de tiempo. Si el intervalo de tiempo es medible y real, la tasa que obtenemos se llama tasa promedio (sobre ese intervalo de tiempo), como se muestra en la gráfica anterior. Si imaginamos que el intervalo de tiempo cae a 0, obtenemos la tasa instantánea, que es la pendiente de la tangente a la curva de concentración versus tiempo en un momento dado (más cálculo). La velocidad al inicio de la reacción se puede obtener tomando la tangente al inicio de la reacción (\(t = 0\)). Esta velocidad inicial es útil en muchas situaciones porque a medida que los reactivos forman productos, estos productos pueden interferir con o inhibir la reacción directa. Esto es particularmente cierto en sistemas biológicos, donde un producto puede influir en su propia formación. Por ejemplo, puede unirse a un sitio en la enzima que cataliza la reacción. Este tipo de interacción es común, y a menudo inhibe la actividad de la enzima (una forma de regulación por retroalimentación).
Podemos medir la velocidad inicial de una reacción usando diferentes concentraciones iniciales de reactivos. Usando un diseño experimental apropiado, podemos averiguar cómo varía la velocidad de reacción con cada reactivo. Para muchas reacciones comunes, la relación entre la tasa y la concentración es bastante sencilla. Por ejemplo, en la reacción:\[\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}+{ }^{-} \mathrm{OH}+\mathrm{Na}^{+} \rightleftarrows\left(\mathrm{CH}_{3}\right)_{3} \mathrm{COH}+\mathrm{Br}^{-}+\mathrm{Na}^{+}\]
la velocidad depende únicamente de la concentración de bromuro de t-butilo\(\left[\left(\mathrm{CH}_{3}\right)_{3}\mathrm{CBr}\right]\), no de la concentración del ión sodio\(\left[\mathrm{Na}^{+}\right]\) o del ion hidróxido\(\left[{}^{-}\mathrm{OH}\right]\). “Pero, ¿por qué solo el bromuro de t-butilo?” bien podrías preguntar. En breve llegaremos a ese punto, porque nos da algunas ideas muy interesantes e importantes sobre el mecanismo de reacción. Primero, profundicemos en un poco más de fondo.
Debido a que la tasa es directamente proporcional a la\(\left[(\mathrm{CH}_{3})_{3}\mathrm{CBr}\right]\), podemos escribir la relación entre tasa y concentración como:\(\text {rate } \propto \left[\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\right]\), o podemos poner en una constante (\(k\)) para hacer la ecuación:\[\text { rate }=k\left[\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\right]\]
También podríamos escribir\[-\Delta\left[\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\right] / \Delta \mathrm{t}=k\left[\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\right] ,\]
o si dejamos que el intervalo de tiempo caiga a cero,\[-d\left[\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\right] / dt=k\left[\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\right] .\]
En todas estas formas, la ecuación se conoce como la ecuación de velocidad para la reacción. La ecuación de tasa debe ser determinada experimentalmente. Vale la pena señalar que no se puede anotar la ecuación de tasa con sólo considerar la ecuación de reacción. (Obviamente, en este caso,\({}^{-}\mathrm{OH}\) o\(\mathrm{Na}^{+}\) no aparecen en la ecuación de tasa.) La constante (\(k\)) se conoce como constante de velocidad y es completamente diferente de la constante de equilibrio (\(\mathrm{K}_{eq}\)). El hecho de que ambos estén designados por\(k\) (una minúscula y una mayúscula) es solo una de esas cosas que tenemos que anotar y asegurarnos de no confundir. Una ecuación de tasa que solo contiene una concentración se denomina ecuación de tasa de primer orden, y las unidades de la constante de velocidad son 1/tiempo.
Ahora, en contraste con la reacción de primer orden del bromuro de metilo y el hidróxido, comparemos la reacción del bromuro de metilo con el hidróxido: [8]\[\mathrm{CH}_{3} \mathrm{Br}+{ }^{-} \mathrm{OH}+\mathrm{Na}^{+} \rightleftarrows \mathrm{CH}_{3} \mathrm{OH}+\mathrm{Br}^{-}+\mathrm{Na}^{+} ,\]
A todos los efectos, esta reacción parece ser exactamente la misma que la comentada en la página anterior. Es decir, el bromo que estaba unido a un carbono ha sido sustituido por el oxígeno del hidróxido. [9] Sin embargo, si realizamos los experimentos, encontramos que la velocidad de reacción depende tanto de la concentración\(\left[\mathrm{CH}_{3}\mathrm{Br}\right]\) de bromuro de metilo como de la concentración de hidróxido\(\left[{}^{-}\mathrm{OH}\right]\). La ecuación de tasa es igual a\(\mathrm{k}\left[\mathrm{CH}_{3} \mathrm{Br}\right]\left[^{-}\mathrm{OH}\right]\). ¿Cómo puede ser esto? ¿Por qué la diferencia? Bueno, lo primero que nos dice es que algo diferente está sucediendo a nivel molecular; los mecanismos de estas reacciones son diferentes.
Las reacciones que dependen de las concentraciones de dos reactivos diferentes se denominan reacciones de segundo orden, y las unidades de\(k\) son diferentes (se puede averiguar cuáles son por análisis dimensional). En general:\ [\ begin {aligned}
&\ text {rate} = k [\ mathrm {A}]\ text {first order}\\
&\ text {rate} =k [\ mathrm {A}] [\ mathrm {B}]\ text {segundo orden (primer orden en}\ mathrm {~A}\ text {y primer orden en}\ mathrm {~B})\
&\ text {tasa} =k [\ mathrm {A}] ^ {2}\ text {segundo orden (en}\ mathrm {~A})\\
&\ text {rate} =k [\ mathrm {A}] ^ {2} [\ mathrm {B}]\ text {tercer orden (segundo orden en}\ mathrm {~A}\ text {y primer orden en}\ mathrm {~B}). \\
\ fin {alineado}\]
Existen varios métodos para determinar la ecuación de velocidad para una reacción. Aquí consideraremos sólo dos. Un método es conocido como el método de las tasas iniciales. La velocidad inicial de la reacción se determina para diversas concentraciones de partida diferentes de reactivos. Claramente, el diseño experimental es de suma importancia aquí. Digamos que está investigando nuestra reacción\(\mathrm{A} + \mathrm{~B} \rightleftarrows 2\mathrm{AB}\). La tasa puede depender de\([\mathrm{A}]\) y/o\([\mathrm{B}]\). Por lo tanto, las concentraciones iniciales de\([\mathrm{A}]\) y\([\mathrm{B}]\) deben ser cuidadosamente controladas. Si\([\mathrm{A}]\) se cambia en un ensayo de reacción, entonces se\([\mathrm{B}]\) debe mantener constante, y viceversa (no se pueden cambiar ambas concentraciones al mismo tiempo porque no sabría cómo afecta cada una a la tasa).
El método de tasas iniciales requiere ejecutar el experimento varias veces usando diferentes concentraciones iniciales. Por el contrario, el método gráfico implica determinar la ecuación de velocidad a partir de una sola ejecución de la reacción. Este método requiere la recolección de un conjunto de datos de concentración versus tiempo (los mismos datos que recogerías para determinar las tasas). Idealmente nos gustaría manipular los datos para que podamos obtener una ecuación lineal (\(y = mx + b\)). Por ejemplo, si tenemos un conjunto de datos\([\mathrm{A}]\) versus tiempo para una reacción, y asumimos que la reacción es de primer orden en\(\mathrm{A}\), entonces podemos escribir la ecuación de velocidad como:\(-d[\mathrm{A}] / dt=k[\mathrm{A}]\).
Ahora bien, si separamos las variables\([\mathrm{A}]\) y\(t\) para obtener:\(-d[\mathrm{A}] / [\mathrm{A}] = kt\). Luego podemos integrar la ecuación a lo largo del periodo de tiempo\(t = 0\)\(t = t\) para llegar a:\[\ln [\mathrm{A}]_{t}=-kt+[\mathrm{A}]_{0} .\]
enlace asociado con la ecuación anterior. [10]
Notarás que esta ecuación tiene la forma de una línea recta; si trazamos nuestros datos (\(\ln [\mathrm{A}]\)versus\(t\)) y si la reacción es de primer orden en\([\mathrm{A}]\), entonces deberíamos obtener una línea recta, donde está la pendiente de la línea\(–k\). Podemos hacer un análisis similar para una reacción que podría ser de segundo orden en\([\mathrm{A}]\):\[\text{rate } = k[\mathrm{A}]^{2} .\]
En este caso, podemos manipular la ecuación de tasa e integrarla para dar la ecuación:\[1 /[\mathrm{A}]_{t}=kt+1 /[\mathrm{A}]_{0}\]
Por lo tanto, el trazado\(1/[\mathrm{A}]\) versus\(t\) daría una línea recta, con una pendiente de\(k\), la constante de velocidad. Este método de análisis rápidamente se vuelve demasiado complejo para reacciones con más de un reactivo (en otras palabras, reacciones con tasas que dependen de ambos\([\mathrm{A}]\) y\([\mathrm{B}]\)), ¡pero puedes esperar eso en tus estudios posteriores!
| Orden | Ley de tarifas | Ley de Tasa Integrada | Gráfica para Línea Recta | Pendiente de Línea |
| 0 | \(\text{rate } = k\) | \([\mathrm{A}]_{t} =-kt + [\mathrm{A}]_{0}\) | \([\mathrm{A}]\)vs.\(t\) | \(–k\) |
| 1 | \(\text{rate } = k[\mathrm{A}]\) | \(\ln [\mathrm{A}]_{t}=-kt+[\mathrm{A}]_{0}\) | \(\ln [\mathrm{A}]\)vs.\(t\) | \(–k\) |
| 2 | \(\text{rate } = k[\mathrm{A}]^{2}\) | \(1 /[\mathrm{A}]_{t}=kt+1 /[\mathrm{A}]_{0}\) | \(1/[\mathrm{A}]\)vs.\(t\) | \(k\) |
Los dos enfoques (múltiples corridas con diferentes condiciones iniciales y el método gráfico para encontrar la mejor línea para ajustarse a los datos) nos proporcionan la ley de tarifas. La pregunta es, ¿qué nos dice la ley tarifaria sobre el mecanismo? Volveremos a esta pregunta al final de este capítulo.
Preguntas para responder
- Resulta que la mayoría de las reacciones simples son de primer o segundo orden. ¿Puedes pensar por qué?
- Diseñar un experimento para determinar la ecuación de velocidad para una reacción\(2\mathrm{A} + \mathrm{B} \rightleftarrows \mathrm{C}\). Usando el método de tasas iniciales y una primera ejecución experimental usando\(0.1-\mathrm{M}\) concentraciones de todos los reactivos, describa los otros conjuntos de condiciones que usaría para averiguar cuál es esa ecuación de tasa.
- ¿Cuál es el número mínimo de corridas de la reacción que tendrías que hacer?
- ¿Cómo determinarías la tarifa para cada uno de tus conjuntos de condiciones?
- Ahora imagina que has determinado que esta reacción\(2\mathrm{A} + \mathrm{B} \rightleftarrows \mathrm{C}\) no depende de ello\([\mathrm{B}]\). Esboza un método gráfico que podrías usar para determinar la ecuación de tasa. ¿Qué datos tendrías que recopilar? ¿Qué harías con él?
Preguntas para más tarde
- ¿Por qué crees que es que no podemos simplemente escribir la ecuación de velocidad a partir de la ecuación de reacción?
- ¿Por qué crees que las ecuaciones de tasa más comunes son de segundo orden?
Temperatura y velocidades de reacción
La temperatura es otro factor importante cuando consideramos las velocidades de reacción. Esto tiene sentido si recuerdas que la gran mayoría de las reacciones involucran colisiones y que los efectos de las colisiones están influenciados por la rapidez con que se mueven los objetos colisionantes. Sabemos intuitivamente que calentar las cosas tiende a hacer que las cosas sucedan más rápido. Por ejemplo, si quieres algo para cocinar más rápido lo calientas a una temperatura más alta (y cocinar, como sabemos, es solo una serie de reacciones químicas). ¿Por qué es así? Si consideramos la reacción del hidrógeno y el oxígeno, discutida en el Capítulo\(7\), que es una reacción altamente exotérmica—explosiva, de hecho. Sin embargo, una mezcla de hidrógeno y oxígeno es bastante estable a menos que se suministre energía, ya sea por calentamiento o una chispa de electricidad. Lo mismo ocurre con la madera y el oxígeno molecular. La pregunta es: ¿Para qué se utiliza la chispa inicial de energía?
La respuesta se encuentra dentro de uno de los principios a los que hemos vuelto una y otra vez: Cuando los átomos forman enlaces, el resultado es un sistema más estable, en comparación con la energía de los átomos no enlazados. Pero no todos los vínculos son igualmente estables; algunos son más estables que otros. Sin embargo, siempre se requiere energía para romper un vínculo, cualquier vínculo. Si va a tener lugar una reacción, entonces al menos uno de los enlaces presentes en los reactivos debe romperse, y esto requiere energía.
Imagina dos reactivos acercándose entre sí. A medida que la reacción comienza a ocurrir, lo primero que sucede es que al menos un enlace en una molécula reaccionante debe comenzar a romperse. Es el paso inicial de ruptura parcial del enlace que requiere una entrada de energía del entorno de la molécula, y la cantidad de energía requerida y disponible determinará si ocurre la reacción. Si la cantidad de energía en el ambiente no es suficiente para comenzar la ruptura de enlaces en los reactivos (por ejemplo, en la quema de madera, se requieren grandes cantidades de energía para la ruptura inicial del enlace), entonces la reacción no ocurrirá sin un “empuje” energético. La madera no solo estalla en llamas (al menos a temperaturas estándar), y tampoco los humanos. [11] La reacción de la quema de madera\(\text{wood } + \mathrm{O}_{2} \rightleftarrows \mathrm{H}_{2}\mathrm{O} + \mathrm{CO}_{2}\),, no ocurre en condiciones normales, pero si la temperatura aumenta lo suficiente, se inicia la reacción. Sin embargo, una vez que comienza la reacción, la energía liberada por la formación de nuevos enlaces es suficiente para elevar la temperatura local y llevar a la ruptura de más enlaces, la formación de nuevos y la liberación de más energía. Mientras haya madera y oxígeno disponibles, el sistema se comporta como un bucle de retroalimentación positivo y autosostenido. La reacción se detendrá si uno de los reactivos se agota o se baja la temperatura.
Es la energía de activación asociada a las reacciones la que es responsable de la estabilidad de nuestro mundo. Por ejemplo, vivimos en una atmósfera de\(\sim 20 \%\) oxígeno (\(\mathrm{O}_{2}\)). Hay muchas moléculas en nuestros cuerpos y en nuestro entorno con las que pueden reaccionar\(\mathrm{O}_{2}\). Si no hubiera barreras energéticas para la combustión (es decir, la reacción con\(\mathrm{O}_{2}\)), estallaríamos en llamas. Tristemente, como habrían atestiguado las brujas de Salem y otros (si hubieran podido), elevamos la temperatura y nosotros sí quemamos. Y una vez que empezamos a quemar, es difícil detener la reacción. Como hemos dicho antes, las reacciones de combustión son exotérmicas. Una vez que han producido suficiente energía térmica, la reacción ya no necesita esa chispa. Pero esa chispa inicial necesita la adición de energía (como la que proporciona un detonador) para que ocurran explosiones.
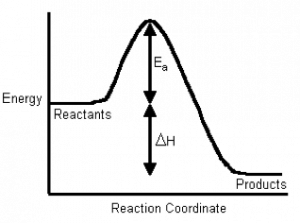
Si trazamos energía versus el progreso de la reacción, podemos obtener una imagen de los cambios de energía que ocurren durante la reacción. Recuerde que la coordenada de reacción en el eje x no es tiempo; hemos visto que las reacciones van hacia atrás y hacia adelante todo el tiempo. Para una reacción simple de un solo paso como se muestra en la figura, el punto más alto en el perfil de energía se denomina estado de transición. No es una entidad estable y solo existe en la escala de tiempo de las vibraciones moleculares (femtosegundos). El cambio de energía entre los reactivos y el estado de transición se llama energía de activación. Esta es la energía que se debe suministrar a los reactivos antes de que pueda ocurrir la reacción. Esta barrera energética de activación es la razón por la que, por ejemplo, podemos mezclar hidrógeno y oxígeno y no explotarán hasta que suministremos una chispa, y por qué podemos bombear gasolina en una atmósfera que contenga oxígeno, aunque sabemos que la gasolina y el oxígeno también pueden explotar. La cantidad de energía que se debe suministrar para producir una reacción es función del tipo de reacción, algunas reacciones (ácido-base) tienen bajas energías de activación y correspondientemente altas tasas, y algunas (oxidación) tienen altas energías de activación y bajas velocidades.
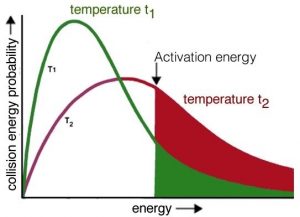
Ahora debería ser más fácil entender cómo el aumento de la temperatura aumenta la velocidad de reacción, al aumentar la energía cinética promedio de las moléculas en el ambiente. Recordemos que aunque las moléculas individuales tienen diferentes energías cinéticas, todas las diferentes poblaciones de moléculas en un sistema tienen la misma energía cinética promedio. Si consideramos el efecto de la temperatura en la distribución Maxwell—Boltzmann de las energías cinéticas, vemos de inmediato que a temperaturas más altas hay relativamente más moléculas con mayor energía cinética. Las colisiones entre estas moléculas de alta energía proporcionan la energía necesaria para superar la barrera de energía de activación, es decir, la energía mínima requerida para iniciar una reacción química. A medida que aumenta la temperatura, aumenta la probabilidad de colisiones productivas entre partículas por unidad de tiempo, aumentando así la velocidad de reacción. Al mismo tiempo, es posible que elevar la temperatura permita que ocurran otras reacciones (quizás reacciones que no hemos estado considerando). Esto es particularmente probable si se trata de mezclas complejas de diferentes tipos de moléculas.
La ecuación de velocidad no parece contener un término para temperatura, y normalmente tenemos que especificar la temperatura a la que se mide la velocidad. Sin embargo, debido a que la velocidad cambia con la temperatura, debe ser la constante de velocidad la que cambia. Efectivamente, se ha determinado experimentalmente que la constante de velocidad es\(k\) puede ser descrita por la ecuación\[k=\mathrm{A}e^{-\mathrm{E}_{a} / \mathrm{RT}} ,\]
donde\(k\) es la constante de velocidad,\(\mahtrm{E_{a}\) es la energía de activación,\(\mathrm{T}\) es la temperatura,\(\mathrm{R}\) y\(\mathrm{A}\) son constantes. [12] Esto se conoce como la ecuación de Arrhenius. Como puede ver,\(k\) es directamente proporcional a la temperatura, e indirectamente proporcional a la energía de activación\(\mathrm{E}_{a}\). La constante a veces\(\mathrm{A}\) se llama factor de frecuencia y tiene que ver con la tasa de colisión. \(\mathrm{A}\)cambia dependiendo del tipo específico de reacción (a diferencia de\(\mathrm{R}\), la constante de gas, que no cambia de reacción a reacción). Una forma de pensar sobre la constante de velocidad es considerarla como una representación de la probabilidad de que una colisión conduzca a productos: cuanto mayor sea la constante de velocidad, más frecuentemente ocurren colisiones productivas y más rápida es la reacción.
La energía de activación para una reacción también depende del tipo de reacción que esté ocurriendo. Por ejemplo, una reacción de base ácida de Brønsted-Lowry tiene una barrera de energía de activación muy baja. En estas reacciones lo único que está sucediendo es que un protón se está transfiriendo de un elemento electronegativo a otro:\[\mathrm{H-Cl}+\mathrm{H-O-H} \rightleftarrows \mathrm{Cl}^{-}+\mathrm{H}_{3} \mathrm{O}^{+}\]
(sacar esto para ver mejor lo que está sucediendo).
La reacción es rápida porque el\(\mathrm{Cl—H}\) enlace es altamente polarizado y débil. En cierto sentido, ya está parcialmente roto. Además, estas reacciones suelen tener lugar en el agua, lo que interactúa y estabiliza las cargas crecientes. Las colisiones de baja energía con moléculas de agua son suficientes para terminar de romper el\(\mathrm{Cl—H}\) enlace. Decimos que reacciones ácido-base como esta se controlan cinéticamente porque ocurren al mezclarse y no requieren calentamiento ni energía extra para continuar. Esencialmente todas las colisiones que involucran a la\(\mathrm{HCl}\) molécula proporcionan suficiente energía para romper el\(\mathrm{H—Cl}\) enlace. Esto también es cierto para casi todas las reacciones de transferencia de protones. Sin embargo, para la mayoría de los otros tipos de reacciones, simplemente mezclar los reactivos no es suficiente. Se debe suministrar energía al sistema para superar esta barrera energética, o tenemos que esperar mucho tiempo para que ocurra la reacción. De hecho, la mayoría de las reacciones orgánicas (aquellas en las que está involucrado el carbono) son bastante lentas. ¿Por qué la diferencia? La respuesta debe ser razonablemente obvia. Simplemente no hay suficiente energía en la gran mayoría de las colisiones entre moléculas para romper un\(\mathrm{C—H}\),\(\mathrm{C—C}\),\(\mathrm{C—N}\), o\(\mathrm{C—O}\) enlace. Si toma el laboratorio de química orgánica, descubrirá que grandes porciones de tiempo se pasan esperando a medida que las soluciones se calientan para hacer que las reacciones sucedan más rápido. Como mencionamos antes, esto es bastante afortunado. Como mencionamos antes, esto es bastante afortunado, ya que estamos (básicamente) organizados por casualidad y selección natural, a partir de colecciones de reacciones orgánicas. Si estas reacciones ocurrieran de manera espontánea y rápida, nos desmoronaríamos y nos acercaríamos al equilibrio (¡y el equilibrio para los seres vivos significa muerte!). Es posible que ya veas el problema potencial en todo esto: generalmente no es recomendable calentar un sistema biológico, pero ciertamente necesitamos sistemas biológicos para sufrir reacciones. Los sistemas biológicos necesitan diferentes reacciones para proceder en diferentes lugares y a diferentes velocidades, sin calentarse. Para ello, los sistemas biológicos (y muchos otros tipos de sistemas) utilizan una amplia gama de catalizadores, tema de nuestra siguiente sección.
Preguntas a Responder:
- Cuando una reacción libera energía, ¿de dónde viene la energía?
- Existe una regla empírica que aumentar la temperatura por\(10^{\circ}\mathrm{C}\) duplicará la velocidad para muchas reacciones.
- ¿Qué factor en la ecuación de Arrhenius siempre está cambiando?
- Explique por qué la velocidad de reacción aumenta cuando aumenta la temperatura.