
4.2: Día 28- Entropía, Energía Libre de Gibbs
- Page ID
- 78960

25
Día 28: Entropía, Energía Libre de Gibbs
D28.1 Entropía y Microestados
Un objetivo importante de la termodinámica química es predecir si los reactivos se cambian a productos o los productos se cambian por reactivos. Para una reacción dada, tal predicción requiere el conocimiento de condiciones específicas de temperatura y de concentraciones o presiones parciales de reactivos y productos. Cuando los reactivos cambian a productos decimos que la reacción es espontánea, o espontánea en la dirección hacia adelante. Cuando los productos cambian a reactivos, decimos que la reacción no es espontánea, o que es espontánea en la dirección inversa. En este contexto, la palabra “espontáneo” no implica que la reacción sea rápida o lenta, solo que los reactivos cambian a productos. Aunque tarde millones de años para que ocurra un proceso, si hay un cambio general de reactivos a productos llamamos al proceso espontáneo.
Es útil definir términos paralelos que se relacionen con un punto de referencia conveniente: condiciones de estado estándar. Si, cuando todas las sustancias están a la presión de estado estándar de 1 bar o a la concentración de estado estándar de 1 M, los reactivos cambian a productos, llamamos a un producto de reacción favorecido. En contraste, si los productos cambian a reactivos en condiciones de estado estándar, un proceso es favorecido por los reactivos. Es decir, un proceso favorecido por el producto es espontáneo bajo las condiciones específicas de presiones o concentraciones de estado estándar y un proceso favorecido por reactivo no es espontáneo en esas condiciones específicas.
Decidir si un proceso es espontáneo requiere conocer el cambio de entalpía del sistema, pero eso no es suficiente. También requiere conocimiento de cambio en otra propiedad: la entropía. (De manera similar, decidir si un proceso es favorecido por el producto requiere conocer el cambio de entalpía estándar del sistema y el cambio de entropía estándar). El cambio de entropía, Δ S, a temperatura constante se define como:
\[{\Delta}S = \dfrac{q_{\text{rev}}}{T} \nonumber \]
Aquí, q rev es la transferencia de calor de energía para un proceso reversible, un proceso teórico que tiene lugar a una velocidad tan lenta que siempre está en equilibrio y su dirección puede ser cambiada (puede ser “invertida”) por un cambio infinitesimalmente pequeño en algunos condición. Un ejemplo de un proceso reversible es fundir agua a 0 °C y 1 bar, donde el agua líquida y el hielo están en equilibrio. Elevar la temperatura un poquito hace que el hielo se derrita; bajar la temperatura un poquito invierte el proceso, haciendo que el agua líquida se congele.
A escala molecular, la entropía de un sistema puede estar relacionada con el número de microestados posibles (W). Un microestado es una configuración específica de las ubicaciones y energías de los átomos o moléculas que comprenden un sistema. La relación es:
S = k B · ln (W)
donde k B es la constante de Boltzmann con un valor de 1.38×10 −23 J/K.
Similar a la entalpía, el cambio en la entropía para un proceso es la diferencia entre sus valores final (S f) e inicial (S i):
\[{\Delta}S = S_{\text{f}}\;-\;S_{\text{i}} = k_\text{B}\cdot\text{ln}(W_{\text{f}})\;-\;k_\text{B}\cdot\text{ln}(W_{\text{i}}) = k_\text{B}\cdot\text{ln}\left(\dfrac{W_{\text{f}}}{W_{\text{i}}}\right) \nonumber \]
Para procesos que implican un incremento en el número de microestados, W f > W i, la entropía del sistema aumenta, Δ S > 0. Por el contrario, los procesos que reducen el número de microestados, W f < W i, producen una disminución en la entropía del sistema, Δ S < 0.
Consideremos el caso general de un sistema compuesto por partículas de N distribuidas entre n cajas. El número de microestados posibles para dicho sistema es n N. Por ejemplo, supongamos que cuatro átomos, un átomo cada uno de He, Ne, Ar y Kr, se distribuyen entre dos cajas como se ilustra en la Figura 1. Hay 2 4 = 16 microestados diferentes. Los microestados con arreglos de partículas equivalentes (sin considerar identidades de partículas individuales) se agrupan y se denominan distribuciones. La probabilidad de que un sistema exista como una distribución dada es proporcional al número de microestados dentro de la distribución. Debido a que la entropía aumenta logarítmicamente con el número de microestados, la distribución más probable es la de mayor entropía.
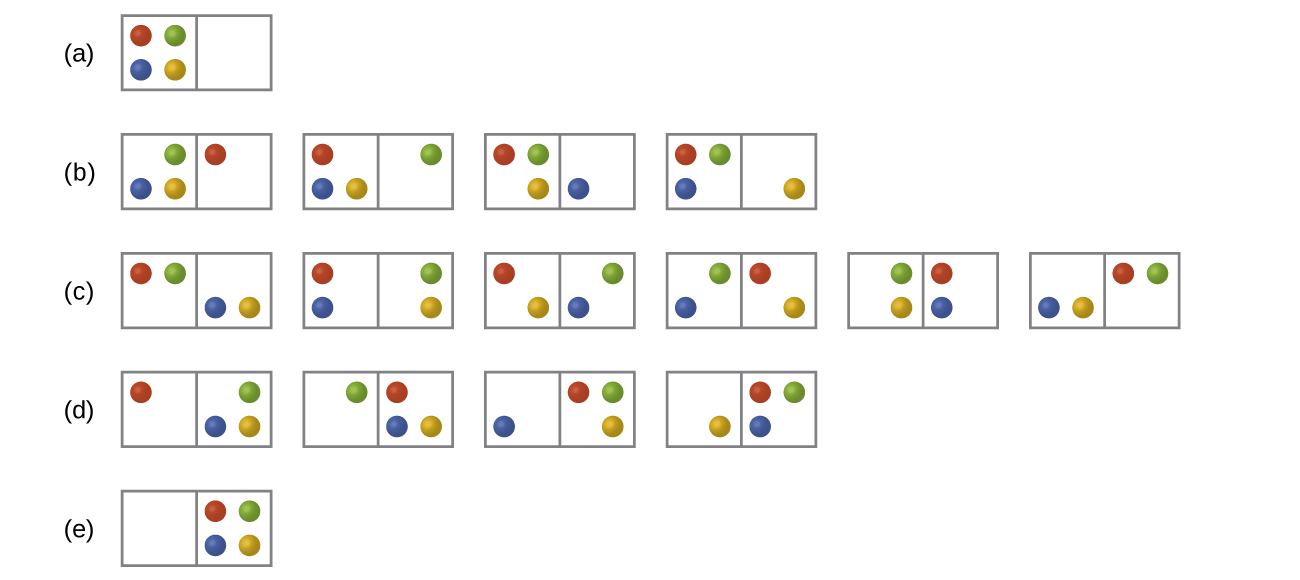
Para el sistema de la Figura 1, la distribución más probable es (c), donde las partículas se distribuyen uniformemente entre las cajas. La probabilidad de encontrar el sistema en esta configuración es o 37.5%. La configuración menos probable del sistema son las distribuciones (a) y (e), donde las cuatro partículas están en una caja, y cada distribución tiene una probabilidad de
.
A medida que agrega más partículas al sistema, el número de microestados posibles aumenta exponencialmente (2 N). Un sistema macroscópico típicamente consiste en moles de partículas (N ≈ 10 23), y el número correspondiente de microestados es asombrosamente enorme. Independientemente del número de partículas en un sistema, las distribuciones con dispersión uniforme de partículas entre las cajas tienen el mayor número de microestados y son las más probables.
Actividad 1: Cambio de entropía y microestados
Considera otro sistema como se muestra en la Figura 2. Este sistema consta de dos objetos, AB y CD, y dos unidades de energía (representadas como “*”). La distribución (a) muestra los tres microestados posibles para el estado inicial del sistema, donde ambas unidades de energía están contenidas dentro del objeto AB caliente. Si se transfiere una unidad de energía, el resultado es la distribución (b) que consta de cuatro microestados. Si se transfieren ambas unidades de energía, el resultado es la distribución (c) que consta de tres microestados.
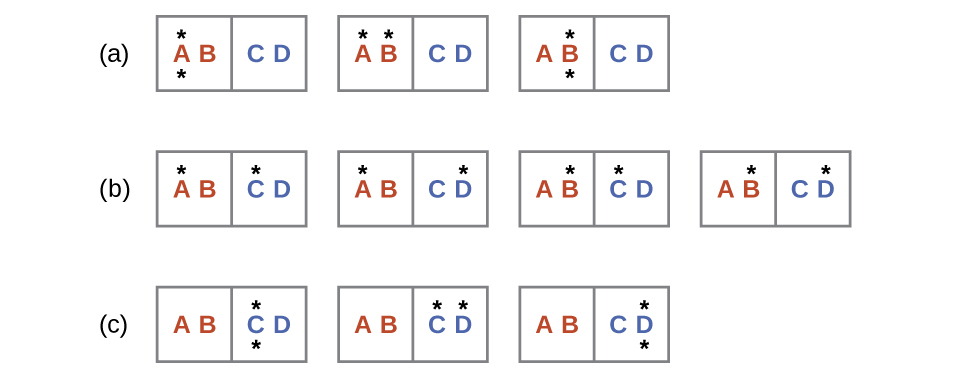
De ahí que podamos describir este sistema por un total de diez microestados. La probabilidad de que no haya transferencia de calor de energía cuando los dos objetos se ponen en contacto (el sistema permanece en distribución (a)) es de 30%. Es mucho más probable que se produzca la transferencia de calor y produzca ya sea la distribución (b) o (c), siendo la probabilidad combinada 70%. El resultado más probable, con 40% de probabilidad, es la transferencia de calor para producir la dispersión uniforme de la energía representada por la distribución (b).
Este ejemplo simple apoya la observación común de que la colocación de objetos fríos y calientes en contacto da como resultado una transferencia de calor que finalmente iguala las temperaturas de los objetos. Tal proceso se caracteriza por un incremento en la entropía del sistema.
D28.2 Predecir el Signo de Δ S
Las relaciones entre entropía, microestados y dispersión materia/energía nos permiten evaluar entropías relativas de sustancias y predecir el signo de cambios de entropía para procesos químicos y físicos.
Considere los cambios de fase ilustrados en la Figura 3. En la fase sólida, las moléculas están restringidas a posiciones casi fijas entre sí y solo oscilan un poco alrededor de estas posiciones; el número de microestados (sólido W) es relativamente pequeño. En la fase líquida, las moléculas pueden moverse una y alrededor de la otra, aunque permanecen relativamente cercanas. Esta mayor libertad de movimiento da como resultado una mayor variación en las posibles ubicaciones de las partículas, W líquido > W sólido. Como resultado, S líquido > S sólido y el proceso de fusión se caracteriza por un incremento en la entropía, Δ S > 0.

Los sólidos y líquidos tienen superficies que definen su volumen, pero en la fase gaseosa las moléculas ocupan todo el contenedor; por lo tanto, para la misma muestra, cada molécula en un gas se puede encontrar en muchas más ubicaciones (y hay muchos más microestados) que en la fase líquida o sólida. En consecuencia, S gas > S líquido > S sólido, y los procesos de vaporización y sublimación implican incrementos en la entropía, Δ S > 0.
Ya hemos discutido que la temperatura de una sustancia es proporcional a la energía cinética promedio de sus partículas. El aumento de la temperatura da como resultado vibraciones más extensas de las partículas en sólidos y movimientos más rápidos de las partículas en líquidos y gases. A temperaturas más altas, la distribución Maxwell-Boltzmann de las energías cinéticas moleculares también es más amplia que a temperaturas más bajas; es decir, existe un mayor rango de energías de las partículas. Así, la entropía para cualquier sustancia aumenta con la temperatura (Figura 4).
La entropía de una sustancia también está influenciada por la estructura de las partículas que la componen. Para las sustancias atómicas, los átomos más pesados poseen mayor entropía a una temperatura dada que los átomos más ligeros. Para los átomos más pesados los niveles de energía correspondientes al movimiento de un lugar a otro están más próximos entre sí, lo que significa que a una temperatura dada hay más niveles de energía ocupados y más microestados. Para las moléculas, un mayor número de átomos (independientemente de sus masas) dentro de una molécula da lugar a más formas en las que la molécula puede vibrar, lo que aumenta el número de posibles microestados. Así, cuantos más átomos haya en una molécula, mayor es la entropía.
Finalmente, las variaciones en los tipos de partículas afectan la entropía de un sistema. En comparación con una sustancia pura, en la que todas las partículas son idénticas, la entropía de una mezcla de diferentes tipos de partículas es mayor debido a las orientaciones e interacciones adicionales que son posibles. Por ejemplo, cuando un sólido se disuelve en un líquido, las partículas del sólido experimentan tanto una mayor libertad de movimiento como interacciones adicionales con las partículas de disolvente. Esto corresponde a una dispersión más uniforme de materia y energía y a un mayor número de microestados. Siendo todas las demás cosas iguales, el proceso de disolución implica por lo tanto un aumento de la entropía, Δ S > 0.
D28.3 Segunda Ley de Termodinámica
La segunda ley de la termodinámica permite predecir si un proceso, como una reacción química, es espontáneo y si el proceso es favorecido por el producto.
En los modelos termodinámicos, el sistema y el entorno lo comprenden todo (es decir, el universo), y así
Δ S univ = Δ S sys + Δ S surr
Para ilustrar esta relación, considere el proceso de transferencia de calor entre dos objetos, uno identificado como el sistema y el otro como el entorno. Hay tres posibilidades para tal proceso:
- Los objetos están a diferentes temperaturas, y la energía se transfiere del objeto más caliente al objeto más frío. Esto siempre se observa que ocurre. Designar el objeto más caliente como el sistema e invocar la definición de entropía produce:
\[{\Delta}S_{\text{sys}} = \dfrac{-q_{\text{rev}}}{T_{\text{sys}}}\;\;\;\;\;\;\;\text{and}\;\;\;\;\;\;\;{\Delta}S_{\text{surr}} = \dfrac{q_{\text{rev}}}{T_{\text{surr}}} \nonumber \]
Los signos aritméticos de q rev denotan la pérdida de energía por el sistema y la ganancia de energía por el entorno. Dado que T sys > T surr en este escenario, Δ S surr es positivo y su magnitud es mayor que la magnitud de Δ S sys. Así, Δ S sys y Δ S surr se suman a un valor positivo para Δ S univ. Este proceso implica un incremento en la entropía del universo.
- Los objetos están a diferentes temperaturas, y la energía se transfiere del objeto más frío al objeto más caliente. Esto nunca se observa que ocurra. Nuevamente designando el objeto más caliente como el sistema:
\[{\Delta}S_{\text{sys}} = \dfrac{q_{\text{rev}}}{T_{\text{sys}}}\;\;\;\;\;\;\;\text{and}\;\;\;\;\;\;\;{\Delta}S_{\text{surr}} = \dfrac{-q_{\text{rev}}}{T_{\text{surr}}} \nonumber \]
La magnitud de Δ S surr es nuevamente mayor que la de Δ S sys, pero en este caso, el signo de Δ S surr es negativo, dando un valor negativo para Δ S univ. Este proceso implica una disminución en la entropía del universo. (Obsérvese también que la posibilidad 1, que es la inversa de este proceso, siempre ocurre.)
- La diferencia de temperatura entre los objetos es infinitesimalmente pequeña, T sys ≈ T surr, por lo que la transferencia de calor es termodinámicamente reversible. En este caso, el sistema y el entorno experimentan cambios de entropía que son iguales en magnitud y por lo tanto se suman a un valor de cero para Δ S univ. Este proceso no implica ningún cambio en la entropía del universo.
Estos resultados conducen a la segunda ley de la termodinámica: todos los cambios que tienen lugar por su propia cuenta (son espontáneos) implican un aumento en la entropía del universo.
| Δ S univ > 0 | espontáneo (se lleva a cabo por su propia voluntad) |
| Δ S univ < 0 | no espontáneo (se produciría una reacción inversa) |
| Δ S univ = 0 | el sistema está en equilibrio |
Para muchas aplicaciones realistas, el entorno es vasto en comparación con el sistema. En tales casos, la transferencia de calor de energía hacia o desde el entorno como resultado de algún proceso es una fracción casi infinitesimal de su energía térmica total. Por ejemplo, la combustión de un combustible hidrocarbonado en el aire implica la transferencia de calor de un sistema (las moléculas de combustible y oxígeno que reaccionan para formar dióxido de carbono y agua) a entornos que son significativamente más masivos (la atmósfera terrestre). Como resultado, q surr es una buena aproximación de q rev, y la segunda ley puede afirmarse como:
Δ S univ = Δ S sys + Δ S surr = Δ S sys + q surr /T
Podemos usar esta ecuación para predecir si un proceso es espontáneo.
Actividad 2: Entalpía, Entropía y Reacciones Espontáneas
D28.4 Tercera Ley de Termodinámica
Considera la entropía de un sólido puro, perfectamente cristalino que no posee energía cinética (es decir, a una temperatura de cero absoluto, 0 K). Este sistema puede ser descrito por un solo microestado, ya que su pureza, perfecta cristalinidad y completa falta de movimiento significa que no hay más que una ubicación posible para cada molécula idéntica que comprende el cristal (W = 1). Según la ecuación de Boltzmann, la entropía de este sistema es cero:
S = k B · ln (W) = k B · ln (1) = 0
Esta condición limitante para la entropía de un sistema representa la tercera ley de la termodinámica: la entropía de una sustancia cristalina pura y perfecta a 0 K es cero.
A partir de entropía cero a cero absoluto, es posible realizar mediciones calorimétricas cuidadosas (q rev /T) para determinar la dependencia de temperatura de la entropía de una sustancia y derivar valores absolutos de entropía a temperaturas más altas. (Nótese que, a diferencia de los valores de entalpía, la tercera ley de la termodinámica identifica un punto cero para la entropía; así, no hay necesidad de entalpías de formación y cada sustancia, incluidos los elementos en sus estados más estables, tiene una entropía absoluta). Los valores de entropía estándar (S°) son las entropías absolutas por mol de sustancia a una presión de 1 bar o una concentración de 1 M. El cambio de entropía estándar (Δ r S°) para cualquier proceso químico puede calcularse a partir de la entropía estándar de sus especies reaccionantes y productos:
Δ r S° = S° (productos) − S° (reactivos)
La tabla de termodinámica en el apéndice enumera entropías estándar de compuestos selectos a 298.15 K.
Supongamos que se produce una reacción química exotérmica a presión atmosférica constante. Hay una transferencia de calor de energía del sistema de reacción al entorno, q surr = — q sys. La transferencia de calor para el sistema es el cambio de entalpía de la reacción debido a que, a presión constante, Δ r H° = q (Sección D27.3). Debido a que la transferencia de energía al entorno es reversible, el cambio de entropía para el entorno también se puede expresar como:
\[{\Delta}S_{\text{surr}} = \dfrac{q_{\text{rev}}}{T} = \dfrac{q_{\text{surr}}}{T} = \dfrac{-q_{\text{sys}}}{T} = \dfrac{-\Delta H_{\text{sys}}}{T} = \dfrac{-\Delta _{\text{r}}H^{\circ}}{T} \nonumber \]
El mismo razonamiento se aplica a una reacción endotérmica: q sys y q surr son iguales pero tienen signo opuesto.
También, para un sistema de reacción química, Δ S sys = Δ r S° (el cambio de entropía estándar para la reacción). Por lo tanto, Δ S univ se puede expresar como:
Δ S univ = Δ S sys + Δ S surr = Δ r S° − Δ r H°/T
La conveniencia de esta ecuación es que, para una reacción dada, Δ S univ puede calcularse a partir de datos termodinámicos para el sistema únicamente. Es decir, de los datos que se encuentran en el Apéndice.
D28.5 Energía Libre de Gibbs
Una nueva propiedad termodinámica fue introducida a finales del siglo XIX por el matemático estadounidense Josiah Willard Gibbs. La propiedad se llama energía libre de Gibbs (G) (o simplemente la energía libre), y se define en términos de entalpía, entropía y temperatura de un sistema:
G sys = H sys − TS sys
El cambio en la energía libre de Gibbs (Δ G) a temperatura constante puede expresarse como:
Δ G sys = Δ H sys − T Δ S sys
Δ G se relaciona con si un proceso es espontáneo. Esta relación se puede ver comparándola con la segunda ley de la termodinámica:
Δ S univ = Δ S sys + Δ S surr
Δ S univ = Δ S sys — ΔH sys /T
Al multiplicar ambos lados de esta ecuación por − T, y reorganizar, se obtiene:
− T Δ S univ = Δ H sys — T Δ S sys
Que se puede comparar con la ecuación:
Δ G sys = Δ H sys − T Δ S sys
Por lo tanto:
Δ G sys = − T Δ S univ
Para un proceso que sea espontáneo, Δ S univ debe ser positivo. Debido a que la temperatura termodinámica es siempre positiva (los valores están en kelvin), Δ G debe ser negativa para un proceso que avanza por su propia cuenta.
| Δ S univ > 0 | Δ G sys < 0 | espontáneo (se lleva a cabo por su propia voluntad) |
| Δ S univ < 0 | Δ G sys > 0 | no espontáneo (se produciría una reacción inversa) |
| Δ S univ = 0 | Δ G sys = 0 | en equilibrio |
D28.6 Cálculo Δ G°
Un enfoque conveniente y común para calcular Δ r G° para reacciones es mediante el uso de datos termodinámicos de estado estándar. Un método implica determinar primero los Δ r H° y Δ r S°, luego usando la ecuación:
Δ r G° = Δ r H° − T Δ r S°
También es posible utilizar la energía de formación libre de Gibbs estándar (Δ f G°) de los reactivos y productos involucrados en la reacción para calcular Δ r G°. Δ f G° es el cambio de energía libre de Gibbs que acompaña a la formación de un mol de una sustancia a partir de sus elementos en sus estados estándar. Es por definición cero para la forma más estable de una sustancia elemental bajo condiciones estatales estándar.
Para una reacción genérica:
m A + n B ⟶ x C + y D
Δ r G° de Δ f G° sería:
Δ r G° = Δ f G° (productos) − f G° (reactivos) = [x Δ f G° (C) + y Δ f G° (D)] — [m Δ f G° (A) + n Δ f G° (B)]
Pregunta Podia
Utilizar los datos del Apéndice para calcular Δ r S° para esta reacción a 298 K:
2 NO (g) + O 2 (g) → 2 NO 2 (g) gas
incoloro gas incoloro → gas rojo-marrón
Predecir si la reacción es favorecida por el producto (espontánea en condiciones de estado estándar) o favorecida por el reactivo (no espontánea en condiciones de estado estándar).
Después de hacer tu predicción, mira este video donde NO (g) se agrega a O 2 (g) en un matraz:
https://mediaspace.wisc.edu/id/1_5g4s60ey
¿El video valida tu predicción? Explique por qué o por qué no.
Si el video no valida tu predicción, trata de averiguar por qué tu predicción no funcionó y vuelve a hacerlo de una manera diferente.
Si el video sí valida tu predicción, explica qué aspecto termodinámico de la reacción da cuenta de las observaciones en el video.
Dos días antes de la próxima sesión de toda la clase, esta pregunta de Podia se pondrá en vivo en Podia, donde podrás enviar tu respuesta.