4.12: Construcción de Estructuras Parciales en Espectroscopia de RMN y Determinación de Estructura Combinada
- Page ID
- 79131
Cuando miras un espectro IR, inmediatamente ves pequeños trozos de la estructura, porque ves vínculos individuales. Usted sabe de un vistazo que el compuesto contiene un enlace C=O o un enlace O-H. Eso puede ser muy tranquilizador, porque rápidamente te imaginas a lo que te estás enfrentando.
Los espectros de RMN a menudo requieren más trabajo. Puede que tengas que poner lápiz a papel para llegar a una estructura. El trabajo vale la pena, porque se puede obtener una imagen mucho más detallada de la estructura.
Empecemos con un espectro de RMN de 13 C. Supongamos que tenías picos en el espectro a 200, 35 y 15 ppm. Podríamos asignar estos tres picos de la siguiente manera:
| turno (ppm) | estructura parcial |
| 200 | sp 2 C =O |
| 35 | sp 3 C -C=O |
| 30 | sp 3 C -C=O |
| 15 | sp 3 C -C |
Estamos diciendo que el primer carbono se encuentra en la sp 2 o región plana trigonal, y que está hasta ahora campo abajo debido a un doble enlace con el oxígeno. Los otros dos carbonos están en la región sp 3 o tetraédrica. Uno de ellos no está muy lejos abajo; probablemente solo esté unido a otro carbono sp 3. Los otros dos, a 35 y 30 ppm, están ambos un poco más abajo. Eso es alrededor del lugar correcto para un carbono tetraédrico unido a un carbono plano trigonal; es decir, estos carbonos están unidos cada uno a un doble enlace o a un carbonilo.
En la estructura parcial, siempre ponemos en negrita o subrayamos el carbono que corresponde al pico que estamos discutiendo. Si no haces eso, no está claro si el pico a 30 proviene de un carbono al lado del carbonilo (C=O), o del carbono en el propio carbonilo. También, en el pico a los 15, queremos dejar claro que estamos hablando de un solo átomo de carbono; dejar la estructura parcial como C-C implica de alguna manera que esta espectroscopia observa enlaces, pero no lo hace. La espectroscopia IR observa enlaces. 13 La espectroscopia de RMN C observa átomos de carbono.
Ahora, supongamos que miramos el espectro de RMN 1H para el mismo compuesto. A lo mejor vemos tres picos esta vez. Hay un cuarteto que integra para 2H a 2.3 ppm, un singlete que integra para 3H a 2.1 ppm y un triplete que integra para 3H a 1.1 ppm. Ingresamos esas características en una tabla. Esta vez, hay tres características que explicar para cada pico.
| turno | integ. | multipl. | estructura parcial |
| 2.3 | 2H | cuarteto | CH 3 -C H 2 -C=O |
| 2.1 | 3H | singlet | C H 3 -C=O |
| 1.1 | 3H | triplete | C H 3 -CH2 |
En primer lugar, tenemos que explicar el turno. Todos estos picos se encuentran en el extremo superior del espectro (por debajo de 5 ppm), por lo que probablemente sean de hidrógenos en carbonos sp 3 o tetraédricos. Los dos primeros están ligeramente abajo, poco más allá de 2 ppm. Eso sugiere que los carbonos sp3 a los que están unidos pueden a su vez estar unidos a los carbonos sp 2: bien dobles enlaces o carbonilos. Ya sabemos que hay un carbonilo del espectro 13 C, así que supongamos que eso es lo que está provocando el desplazamiento cerca de 2 ppm. El tercer pico, a 1.1 ppm, está en el rango normal; este hidrógeno está en un carbono tetraédrico, probablemente unido a otros carbonos tetraédricos.
Para demostrar lo que nos está diciendo la integración, solo mostramos el número correcto de hidrógenos. Hay dos hidrógenos responsables del pico a 2.3 ppm. Otros tres son los responsables del pico a 2.1 ppm, y otros tres dan lugar al pico a 1.1 ppm.
Por último, hay que explicar la muliplicidad. El pico a 2.3 ppm es un cuarteto, por lo que por la regla “n+1" debe estar al lado de un grupo CH 3. El pico a 1.1 ppm es un triplete, por lo que debe estar al lado de un grupo CH 2. (No lleva mucho tiempo darse cuenta de que estos dos picos representan hidrógenos que están uno al lado del otro). Finalmente, el pico a 2.1 ppm es un singlete. No tiene vecinos de hidrógeno en absoluto.
Observe que no necesitamos saber cuál es la estructura para poder rellenar estas estructuras parciales. Solo estamos anotando lo que nos dicen los datos. A partir de ahí, no está muy lejos para determinar la estructura general.
Rellenar estructuras parciales para los siguientes picos.
a) 10.1 ppm, 1H, triplete b) 3.4 ppm, 1H, septeto c) 7.3 ppm, 2H, triplete
d) 5.4 ppm, 1H, cuarteto e) 1.4 ppm, 2H, sexteto f) 8.0 ppm, 1H, singlete
g) 2.1 ppm, 3H, singlete h) 6.8 ppm, 2H, doblete i) 0.9 ppm, 6H, doblete
- Contestar
-
Los picos aromáticos (benceno, etc.) se marcan como “Ar” para distinguirlos de los picos de alqueno que aparecen más arriba (desplazamiento inferior). Además, algunos picos pueden estar en dos posiciones simétricas y están etiquetados con “x2".
a) 10.1 ppm, 1H, triplete, CH 2 -C H =O b) 3.4 ppm, 1H, septeto, O-C H (CH 3) 2
c) 7.3 ppm, 2H, triplete, CH=C H -CH x 2 (Ar) d) 5.4 ppm, 1H, cuarteto, CH 3 -C H =C
e) 1.4 ppm, 2H, sexteto, CH 3 -C H 2 -CH 2 f) 8.0 ppm, 1H, singlete, C=C H-C (Ar)
g) 2.1 ppm, 3H, singlete, C H 3 -C=C o C H 3 -C=O o C H 3 -N; necesita contexto para elegir
h) 6.8 ppm, 2H, doblete, CH=C H -C x 2 (Ar) i) 0.9 ppm, 6H, doblete, CH-C H 3 x 2
Identificar los errores en las siguientes estructuras parciales:
a) 3.6 ppm, 2H, triplete, C H 2 -CH 2 b) 2.1 ppm, 2H, singlete, C H 3 -C=C
c) 7.4 ppm, 2H, doblete, CH=C H 2-C d) 1.8 ppm, 2H, quinteto, C H 2 -CH 4
e) 7.8 ppm, 1H, triplete, -C H =CH 2 f) 1.7 ppm, 1H, nonet, NH 2-C H (CH 3) 2
- Contestar
-
Identificar los errores en las siguientes estructuras parciales:
a) 3.6 ppm, 2H, triplete, C H 2 -CH 2 el primer carbono debe estar unido a O para tener un desplazamiento a 3.6 ppm
b) 2.1 ppm, 2H, singlete, C H 3 -C=C la integral dice solo 2H, no 3H
c) 7.4 ppm, 2H, doblete, CH=C H 2 -C el desplazamiento implica aromático, por lo que solo puede haber un H por carbono; debe haber simetría
d) 1.8 ppm, 2H, quinteto, C H 2 -CH 4 no puede haber cuatro hidrógenos en un carbono; debe haber algunos hidrógenos en cada lado
e) 7.8 ppm, 1H, triplete, -C H =CH 2 el desplazamiento implica aromático, por lo que solo puede haber un H por carbono; debe ser uno por cada lado
f) 1.7 ppm, 1H, nonet, NH 2-C H (CH 3) 2 un nitrógeno unido desplazaría este hidrógeno más allá de 2 ppm; además, el acoplamiento rara vez se ve a través de O o N, por lo que los dos H vecinos a la izquierda probablemente estén en un carbono.
Hay muchas maneras en las que podemos usar la espectroscopia de RMN para analizar compuestos. Una aplicación común es la determinación de una estructura desconocida. Dados los espectros MS, IR, 13C y 1H RMN, ¿cuál podría ser la estructura de una muestra desconocida?
A menudo es más fácil comenzar con el espectro IR.
- identificar al menos tres picos en el espectro IR. ¿Qué picos parecen darte la mayor información de este compuesto?
- no pienses con la cabeza; piensa con las manos. Anota ideas sobre el espectro.
- si está trabajando en un comprobante formal de estructura, en una prueba de clase o un informe de laboratorio, es posible que se le solicite ingresar sus datos en una tabla correlacionando el número de onda con la asignación de picos:
| cm -1 | asst |
Por ejemplo, un estudiante podría obtener el siguiente espectro IR.
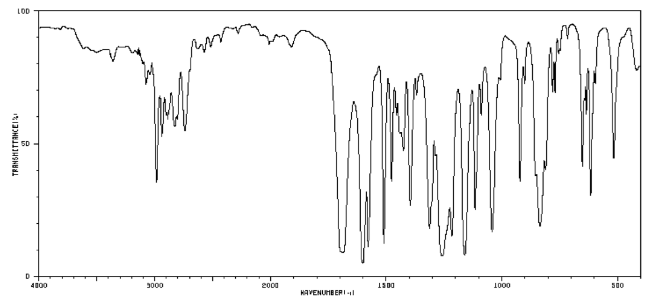
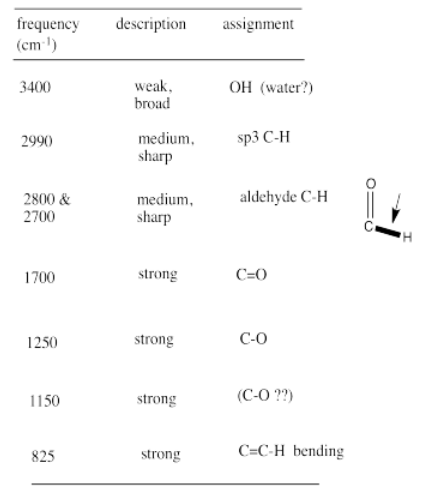
A partir de esa información, construye la siguiente tabla. Incluso podría escribir esta tabla, a mano, directamente en su espectro. Ella hace notas útiles en los bordes, e incluso podría incluir algunas conjeturas, que luego tachó, pero no borra. Se le ayuda en esta tarea consultando una tabla IR, que sugiere lo que podrían significar algunos de estos picos.
Recuerda:
- tomar nota especial de qué átomos están presentes en el compuesto: C, H, N, O...
- también anote sus ideas iniciales sobre grupos funcionales específicos que puedan estar presentes.
- si no está seguro de una tarea, ponga un signo de interrogación al lado para señalar esta incertidumbre.
- es posible que algunos datos deban descartarse más adelante si no son consistentes con otros datos.
Mira el espectro 13 C.
- ¿Cuántos carbonos diferentes hay, en función del número de picos en el espectro? Este es el primer paso para estimar la fórmula molecular.
- ¿Tienes razones para creer que hay simetría en la estructura? ¿En todo el recinto o solo en parte de él? Ajusta el número de carbonos con los que crees que estás tratando.
- Al igual que en la espectroscopia IR, comenzar a asignar picos, ya sea en el espectro o, si es necesario, en una tabla:
| ppm | asst |
Por ejemplo, un estudiante podría obtener el siguiente espectro de RMN de 13 C:
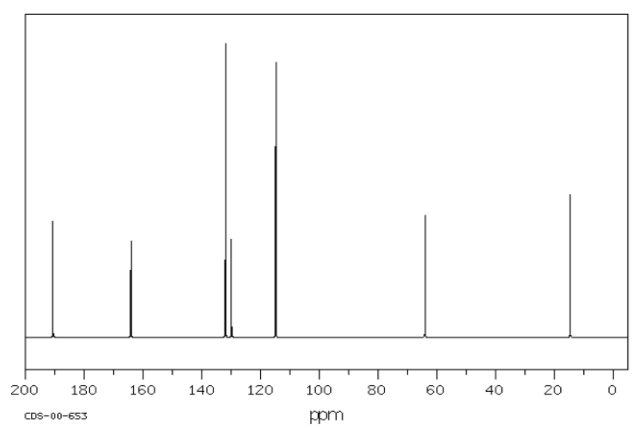
A partir de esa información, reúne la siguiente tabla:
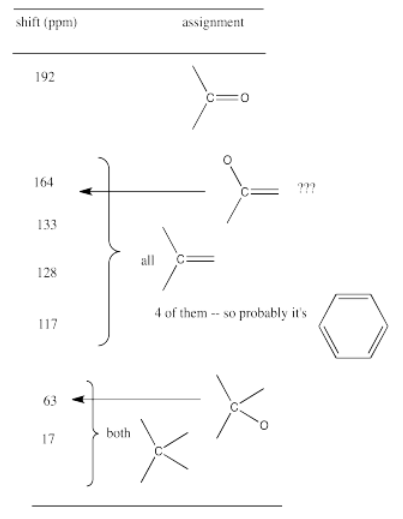
Recuerda:
- podrá asignar todos los picos en el espectro de RMN, no solo unos pocos como en IR.
Al igual que en la RMN de 13 C, debería poder asignar todos los picos en el espectro de RMN 1H. Es posible que puedas hacerlo tomando notas sobre el espectro. Si crees que conoces la estructura, es posible que puedas dibujarla y anotar qué pico pertenece a qué protón.
Una prueba formal de estructura podría requerir una tabla de asignaciones.
|
ppm |
int |
mult |
estructura parcial |
asignación |
- Esta tabla demuestra su capacidad para leer el espectro. ¿Se puede decidir qué proporción de protones sugiere la línea integral? ¿Puedes decidir si un pico es un cuarteto?
- La columna de estructura parcial debe explicar el desplazamiento, integración y multiplicidad para el pico en esa fila. No debe mostrar ninguna otra información de otra parte de la estructura. Esta restricción te obliga a demostrar una comprensión profunda de los datos de una manera que “obtener la respuesta correcta” no lo hace.
- La columna de estructura parcial se rellena mejor con dibujos, no palabras. El dibujo es una estructura parcial.
- Debido a que la estructura parcial mostrará los protones absorbiendo en el turno de esa fila así como los protones vecinos, es necesario distinguir entre ellos en su imagen. La mayoría de la gente da un círculo o subraya o pone en negrita los protones que aparecen en el turno dado en esa fila.
- Cuando termine con la columna de estructura parcial, debería poder vincular las estructuras parciales para hacer una estructura completa en la columna de asignación.
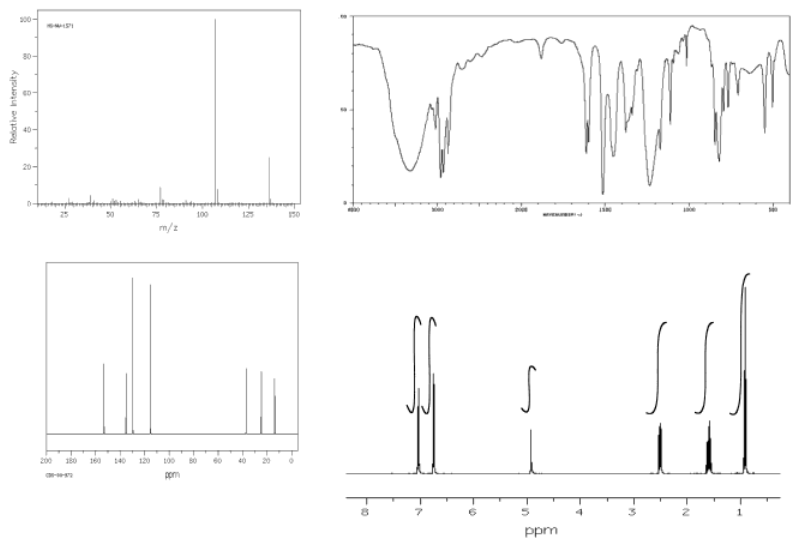
A continuación se da un ejemplo de un espectro y su tabla de datos que lo acompaña. Aquí está el espectro:
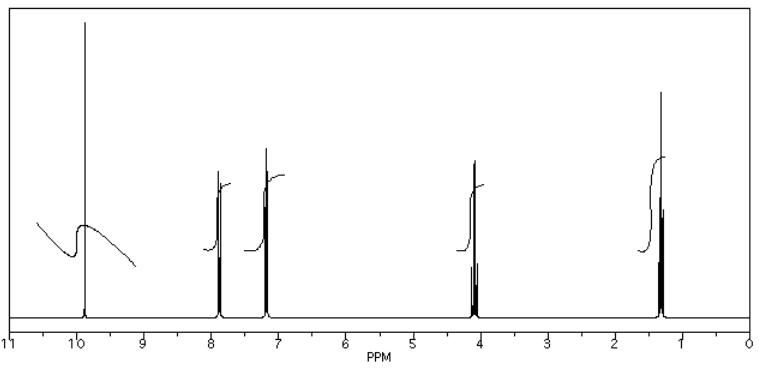
Aquí hay una tabla de datos:
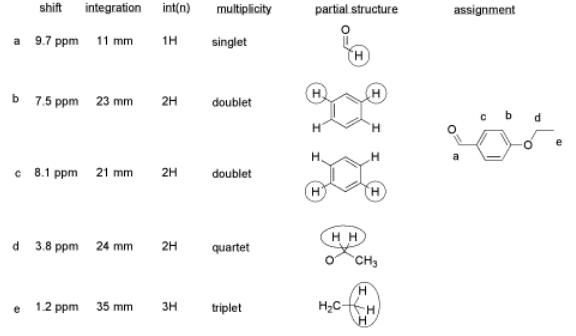
Cosas a tener en cuenta:
- Este alumno ha utilizado dos columnas de integración en lugar de solo una.
- La primera columna muestra la integral medida a partir del espectro. Probablemente usó una regla.
- La segunda columna, a la que llamó int (n), contiene una relación conveniente tomada de los datos brutos. Esta proporción es más fácil de usar en sus asignaciones.
- También tenga en cuenta que el pico a 9.7 ppm no tiene una muy buena integral. Aquí hay un problema de “fase” o de “nivel e inclinación” que se puede corregir usando el software de RMN, pero esto a veces es difícil de hacer. Si hubiera tomado una impresión automática de esta medida integral, habría obtenido un número extraño; en este caso, sería alrededor de -5, porque el final de la línea integral es menor que el inicio. Sin embargo, claramente no es un número negativo de hidrógenos. En cambio, ha medido el ascenso vertical en la integral y lo ha registrado; no es perfecto, sino que es una estimación justa en este caso.
Hay un par de herramientas adicionales que pueden ayudar a confirmar la estructura en este punto. Alternativamente, si la estructura sigue siendo esquiva, estas herramientas podrían ayudar a producir algunas ideas.
La primera herramienta es la fórmula. Una vez que tenemos tablas de RMN, podemos comenzar a adivinar los números de carbonos e hidrógenos en la estructura. Con la adición de una tabla IR, podemos comenzar a adivinar la presencia de otros átomos, como el oxígeno o tal vez nitrógeno.
Por ejemplo, en la tabla de RMN de 13 C anterior, hubo siete picos. Eso significa que probablemente haya al menos siete carbonos. Podemos comenzar la fórmula molecular como C 7. Sin embargo, puede haber carbonos adicionales si hay alguna simetría. También puede haber algunos carbonos que no aparecen muy bien en el espectro. Si alguna vez has obtenido un verdadero espectro de RMN de 13 C, sabrás que los picos de carbonilo pueden ser difíciles de encontrar, especialmente si no hay hidrógenos unidos al carbonilo. En la tabla anterior, parecía que había un benceno, así que tal vez había realmente seis carbonos en la región aromática, y no sólo cuatro carbonos. Eso significaría que la fórmula, hasta el momento, es C 9.
En la tabla de RMN de 1H, las integrales sumaron hasta un total de 10H. Entonces, tal vez la fórmula es C 9 H 10.
Además, la tabla IR sugiere la posible presencia de dos átomos de oxígeno diferentes. La fórmula en realidad puede ser C 9 H 10 O 2.
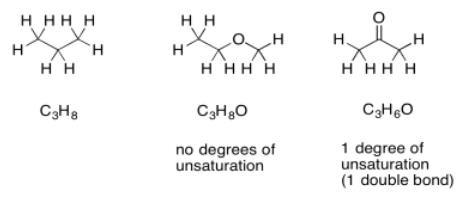
Una vez que tenemos una fórmula, en realidad obtenemos una gran cantidad de información automáticamente. Una de las piezas más importantes es “unidades de insaturación” o “grados de insaturación” (DU). El DU es el resultado de una comparación formal de la relación C/H en el compuesto con la de un alcano normal. En un alcano normal, la fórmula es siempre C n H 2n+2. Si imaginas una cadena larga de hidrocarburos, habrá dos hidrógenos en cada carbono a lo largo de la cadena, más uno más hidrógeno en cada extremo de la cadena. Sin embargo, un alqueno contiene un enlace pi, y en el sitio de ese enlace pi hay dos átomos de hidrógeno que faltan en esa fórmula alcano. Un alqueno simple siempre tiene la fórmula C n H 2n. Ese par faltante de hidrógenos en la fórmula se llama grado de instauración.
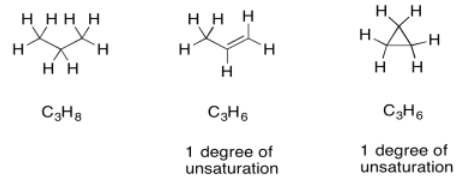
Lo mismo le pasa también a la fórmula si hay un anillo presente. Una DU puede corresponder a la presencia de un doble enlace o un anillo. Si DU=2, puede haber dos dobles enlaces, dos anillos, o uno de cada uno.
Si hay átomos de oxígeno presentes en la fórmula, podemos simplemente ignorarlos y prestar atención a la parte hidrocarbonada. Conceptualmente, debido a que el oxígeno forma dos enlaces, podemos pensar que se aprieta entre dos átomos cualesquiera en una estructura hidrocarbonada para formar un nuevo compuesto. La relación de carbono a hidrógeno no cambia. Si hay un grado de desesturación en una fórmula que contiene oxígeno, simplemente sugiere la presencia de un anillo o un doble enlace, al igual que en un hidrocarburo.
En ocasiones, si hay otros átomos presentes, necesitamos ajustar la fórmula para tomarlos en cuenta. Por ejemplo, cada vez que se encuentra un halógeno en la estructura, reemplaza conceptualmente a un átomo de hidrógeno. Para que se encuentre un halógeno en la estructura, tendría que haber uno menos átomos de hidrógeno para abrir una mancha para el halógeno. Para ajustar la presencia de un halógeno, necesitamos agregar un hidrógeno a la fórmula, luego compararlo con la fórmula de alcano estándar.
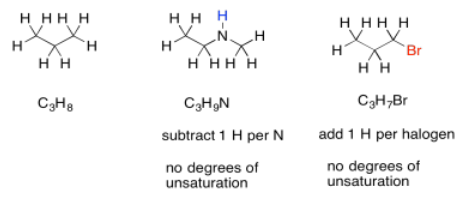
El nitrógeno, por otro lado, tiene tres enlaces. A diferencia del oxígeno, si lo apretamos entre otros dos átomos, todavía necesita un enlace extra. Siempre aporta un hidrógeno extra a la fórmula. Para ajustar la presencia de nitrógeno, necesitamos restar un H de la fórmula, luego compararlo con la fórmula alcano estándar.
En la fórmula que acabamos de calcular, tenemos C 9 H 10 O 2. Podemos ignorar los oxígenos y mirar el C9H10. Si éste fuera un hidrocarburo saturado con nueve carbonos, su fórmula sería C 9 H 20 (ya que 2 x 9 + 2 = 20). Nos faltan cinco pares de hidrógenos, por lo que DU = 5. Eso es mucho. Sin embargo, si tenemos un benceno en la estructura, eso representaría tres dobles enlaces y un anillo todos a la vez. Que cuatro grados de saturación. Un carbonilo adicional elevaría el número hasta los cinco requeridos. Si aún no hubiéramos llegado a la idea de un anillo de benceno, esta comparación podría hacernos pensar en ello. Alternativamente, si supiéramos del benceno pero aún no hubiéramos manchado el carbonilo, podríamos estar al pendiente de él ahora.
Una vez que tenemos una posible fórmula, otra herramienta útil es la espectrometría de masas (EM). Aunque no sepas mucho de espectrometría de masas, la idea básica es sencilla. Un espectrómetro de masas toma una molécula y la golpea en pequeños pedazos, luego mide los pesos moleculares de cada uno de esos fragmentos. Si tienes suerte cuando ejecutas el experimento, algunas de las moléculas quedan intactas, y también obtienes el peso molecular de toda la molécula.
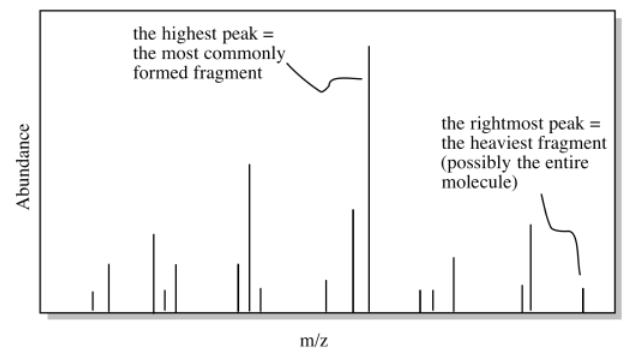
Si calculamos el peso molecular basado en la fórmula y lo comparamos con el posible peso molecular del espectro de masas, podríamos obtener confirmación de que estamos en el camino correcto. Alternativamente, tal vez nuestro peso molecular calculado se quede corto. Si estamos fuera por 16, tal vez nos hayamos perdido un átomo de oxígeno en alguna parte. Si estamos fuera por 14, tal vez nos hemos perdido un carbono y un par de hidrógenos. Esta información podría ayudarnos a corregir algunos errores.
En el ejemplo anterior, la fórmula conduce a un peso molecular de 150 g/mol. Si el espectro de masas no coincidía, nos gustaría revisar nuestro trabajo para ver si pasamos por alto algo.
Usando el enfoque descrito anteriormente, construir un caso para la estructura del compuesto representado por los datos a continuación.