
13.6.1: Alquílicos metálicos
- Page ID
- 81110

Parte 1:
Con este post finalmente llegamos a los ligandos definitorios de la química organometálica, los alquilos. Los alquilos metálicos presentan un enlace σ metal-carbono y suelen ser ligandos actores, aunque algunos ligandos de alquilo se comportan como espectadores. Nuestro objetivo será comprender la dependencia general del comportamiento de los ligandos alquílicos sobre el centro metálico y los sustituyentes del ligando. Usando este conocimiento, podemos hacer comparaciones significativas entre complejos de alquilo de metal relacionados y predicciones educadas sobre su probable comportamiento. Debido a que los ligandos de alquilo son fundamentales para la química organometálica, he decidido difundir esta discusión a través de múltiples publicaciones. Primero nos ocuparemos de las propiedades generales de los alquilos metálicos.
En la serie Simplifying the Organometallic Complex, descompusimos el enlace M-C en un metal cargado positivamente y carbono cargado negativamente. Este procedimiento de deconstrucción es consistente con las electronegatividades relativas del carbono y los metales de transición. Puede ser muy útil para nosotros imaginar alquilos metálicos esencialmente como carbaniones estabilizados, pero también es importante entender que los enlaces M-C abarcan desde extremadamente iónicos y similares a sales (nACh 3) hasta esencialmente covalentes ([hGCH 3] +). La reactividad del ligando alquílico está inversamente relacionada con la electronegatividad del centro metálico.
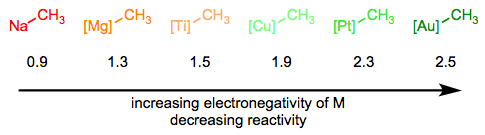
La hibridación del átomo de carbono también es importante, y la tendencia aquí sigue la tendencia en la nucleofilia en función de la hibridación en química orgánica. Los ligandos sp - hibridados son los menos nucleofílicos, seguidos de los ligandos sp 2 y sp 3 respectivamente.
La historia de los alquilos de metales de transición es un ejemplo intrigante de un paradigma científico incorrecto. Después de varios intentos infructuosos de aislar alquilos metálicos estables, los químicos organometálicos en la década de 1920 tuvieron la idea de que los enlaces metal-carbono eran débiles en general. Sin embargo, estudios posteriores mostraron que fue la inestabilidad cinética, no termodinámica, la que tuvo la culpa de nuestra incapacidad para aislar alquilos metálicos. En otras palabras, la mayoría de los alquilos metálicos son susceptibles a vías de descomposición con barreras de activación bajas; la inestabilidad del enlace M-C per se no es la culpable. Crabtree cita valores típicos de 30-65 kcal/mol para las resistencias de unión M-C.
¿Cuáles son las principales vías de descomposición de los complejos de alquilo metálico? La eliminación de β-hidruro es la más común. Termodinámicamente, la ubicuidad de la eliminación de β-hidruro hace que los enlaces senso-M—C corran 30-65 kcal/mol, mientras que los enlaces M—H tienden a ser más fuertes. La siguiente figura resume el mecanismo aceptado y los requisitos de eliminación de β-hidruro. Revisaremos esta reacción fundamental de los complejos organometálicos en un futuro post.
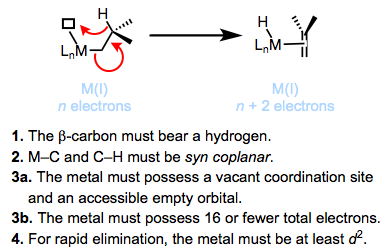
Los complejos de alquilo metálico cinéticamente estables violan uno de los requisitos para la eliminación de β-hidruro. Los complejos de metilo y neopentilo carecen de β-hidrógenos, violando el requisito 1. Se pueden usar ligandos quelantes de unión estrecha para evitar la formación de un sitio de coordinación vacío, violando los requisitos 3a y 3b. Se conocen complejos de titanio que violan el requisito 4 y eliminan solo muy lentamente; se requiere la donación inversa del metal al σ* C—H para una rápida eliminación (ver más abajo).
La eliminación reductiva es una segunda vía de descomposición común. El ligando de alquilo se engancha con un segundo ligando tipo X en el metal, y el metal se reduce en dos unidades con una disminución en el recuento total de electrones en dos unidades. He omitido flechas curvas aquí porque se conocen diferentes mecanismos de eliminación reductiva. Discutiremos los requisitos de eliminación reductiva en detalle en una publicación futura; por ahora, es importante señalar que la estabilidad termodinámica de C—X versus la de (M—X + M—C) es una fuerza impulsora crítica para la reacción.
Cuando X = H, la eliminación reductiva es casi siempre termodinámicamente favorable; por lo tanto, los complejos aislables de hidruro de alquilo son raros. Este comportamiento es una característica, no un error, cuando consideramos que la química de hidrogenación depende de ello! Por otro lado, cuando X = halógeno la eliminación reductora suele ser desfavorecida. La eliminación reductiva de C—C (X = C) se puede favorecer termodinámicamente, pero suele ser más lenta que la correspondiente eliminación de C—H.
Los complejos que no pueden someterse a la eliminación de β-hidruro son a veces susceptibles a la eliminación α-eliminación, un proceso mecanísticamente similar que forma un carbeno metálico. Este proceso es particularmente fácil cuando la posición α es activada por un donante de electrones adyacente (los carbenos Fischer son el resultado).
En algunos complejos de alquilo metálico, los enlaces C—H en las posiciones α, β o incluso más lejanas pueden servir como donantes de electrones al centro metálico. Esta idea está respaldada por la evidencia cristalográfica y los cambios químicos de RMN (los hidrógenos donadores cambian a campo alto). Tales interacciones se llaman interacciones agósticas, y se asemejan a un estado de transición “interrumpido” para la eliminación de hidruro. Los complejos alquílicos que no pueden sufrir eliminación de β-hidruro por razones electrónicas (alto estado de oxidación, metales d 0) y los complejos vinílicos comúnmente exhiben este fenómeno. El hecho de que la eliminación de β-hidruro sea lenta para los metales d 0, en su lugar se observan interacciones agósticas, sugiere que la retrodonación de una órbita metálica llena a la σ* C—H es importante para la eliminación de β-hidruro. Aquí hay una interesante revisión reciente de las interacciones agósticas.
En el siguiente post de esta serie, exploraremos con más detalle la síntesis de complejos de alquilo metálico, aclarando particularmente la pregunta: ¿cómo podemos conquistar la eliminación de β-hidruro?
Parte 2:
En este post, exploraremos los métodos sintéticos más comunes para la síntesis de complejos alquílicos. Además de enumerar las reacciones que producen complejos alquílicos, este post también describirá estrategias para evitar la eliminación de β-hidruro cuando los complejos alquílicos aislables son el objetivo. ¡Aquí vamos!
Propiedades de Complejos Alquílicos Estables
Los complejos alquílicos estables deben ser resistentes a la eliminación de β-hidruro. En el último post identificamos cuatro condiciones clave necesarias para que se produzca la eliminación:
1. El β-carbono debe llevar un hidrógeno.
2. Los enlaces M—C y C—H deben ser capaces de lograr una orientación coplanar syn (apuntando en la misma dirección en planos paralelos).
3. El metal debe portar 16 electrones totales o menos y poseer un sitio de coordinación abierto.
4. El metal debe ser al menos d 2.
Los complejos alquílicos estables deben violar al menos una de estas condiciones. Por ejemplo, los complejos de titanio (IV) que carecen de electrones d β-eliminan muy lentamente. El complejo a continuación probablemente también se beneficie de la quelación (ver más abajo).
Se han ideado complejos que son incapaces de lograr la orientación sin-coplanaria requerida para la eliminación, o que carecen de β-hidrógenos por completo. A continuación se ofrecen algunos ejemplos: hay que admirar la astucia de los investigadores que idearon estos complejos. ¡El metalaciclobutano es particularmente llamativo!
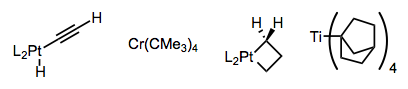
Usando ligandos quelantes de unión estrecha o un grupo director sobre el sustrato, se puede desalentar la formación de complejos de 16 electrones susceptibles a la eliminación de β-hidruro. Observe cómo los ligandos L 2 de unión a hidrógenos en el complejo central de abajo sostienen el centro metálico en un agarre mortal.
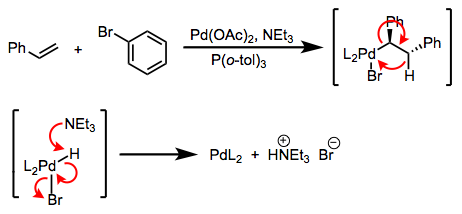
Finalmente, vale la pena señalar que los complejos con un sitio de coordinación abierto, como los complejos de 16 electrones y planares cuadrados de Ni, Pd y Pt importantes para el acoplamiento cruzado, son susceptibles a reacciones con solventes u otras especies en el sitio abierto. Ligandos alquílicos voluminosos ayudan a prevenir estas reacciones secundarias. En el siguiente ejemplo, los grupos metilo de los ligandos o-tolilo se extienden hacia el espacio por encima y por debajo del plano cuadrado, desalentando la aproximación de moléculas de disolvente perpendiculares al plano.
Muchos complejos de metales de transición catalizan la isomerización (E)/(Z) y la isomerización de alquenos terminales (α-olefinas) a isómeros internos mediante la eliminación de β-hidruro. Esto es un testimonio de la importancia de este proceso para los complejos alquílicos. Por supuesto, los complejos alquílicos transitorios pueden parecer susceptibles a la eliminación de β-hidruro, pero si otros procesos son más rápidos, la eliminación no ocurrirá. Así, la optimización de muchas reacciones que involucran complejos alquílicos como intermedios ha implicado acelerar otros procesos a expensas de la eliminación de β-hidruro, la hidrocianación es un buen ejemplo.
Síntesis de Complejos Alquílicos
Los métodos sintéticos dominantes para los complejos alquílicos se basan en el ataque nucleofílico, el ataque electrófilo, la adición oxidativa y la inserción migratoria. Los dos primeros métodos deben ser intuitivos para el químico orgánico; los dos segundos se basan en reacciones más esotéricas (pero no menos importantes) de los complejos organometálicos.
Los metales que llevan buenos grupos salientes son análogos a los electrófilos orgánicos, y son susceptibles al ataque nucleofílico por organolitios, reactivos de Grignard y otros organometálicos polarizados. Estas reacciones pueden verse como una especie de transmetalación, ya que el ligando de alquilo se mueve de un metal a otro. Los ligandos de tipo X aceptores de electrones como —Cl y —Br deberían saltar como buenos grupos de salida. Por otro lado, la sustitución limpia de ligandos tipo L por nucleófilos aniónicos es mucho más rara (resultarían complejos aniónicos).
Muchos complejos metálicos aniónicos son lo suficientemente nucleófilos como para atacar fuentes electrófilas de carbono tales como haluros de alquilo y acilo en un modo de ataque electrófilo. Un par solitario disponible en el sitio de coordinación de metal y abierto son requisitos previos para esta química. La carga en el complejo aumenta en una unidad (en efecto, la carga negativa se transfiere al grupo de salida del electrófilo). Podemos clasificarlas como reacciones de ligación oxidativa; observe que el estado de oxidación del metal aumenta en dos unidades.
La adición oxidativa da como resultado la escisión de un enlace W—Z y la colocación de dos nuevos ligandos tipo X (—W y —Z) en el centro metálico, con un incremento en el estado de oxidación del metal y el recuento total de electrones en dos unidades. Los haluros orgánicos son sustratos extremadamente comunes para esta reacción, el primer paso en el mecanismo de las reacciones de acoplamiento cruzado. El complejo metálico oxidado que contiene nuevos ligandos de alquilo y haluro es el producto final. Observe que se requieren dos sitios de coordinación abiertos (no necesariamente simultáneamente), el centro metálico debe ser susceptible de oxidación de dos electrones, y el número de electrones totales del complejo aumenta en dos. En esencia, los electrones del enlace W—Z se unen al partido del complejo. ¡Toma nota que hay muchos mecanismos conocidos para la adición oxidativa! Exploraremos estos diferentes mecanismos en detalle en una publicación futura.
Finalmente, la inserción migratoria de compuestos orgánicos insaturados es un método importante para la síntesis de ciertos complejos alquílicos, y un paso importante de reacciones organometálicas que resultan además a través de enlaces π. La inserción migratoria es el reverso microscópico de la eliminación de β-hidruro. Los inteligentes entre ustedes pueden notar que el uso de la inserción migratoria para sintetizar complejos alquílicos parece inconsistente con nuestra observación de que su reverso es ubicuo para los alquilos metálicos, ¿no debería favorecer el equilibrio al complejo de hidruro de olefina? En muchos casos este es el caso; sin embargo, hay algunas excepciones notables. Por ejemplo, los complejos de perfluoroalquilo son excepcionalmente estables (¿por qué?) , por lo que a menudo se favorece la inserción de fluoroalquenos sobre la eliminación.
Como señalamos anteriormente, todavía podemos invocar complejos alquílicos cinéticamente estables como intermedios en reacciones siempre que las etapas posteriores sean más rápidas. En el siguiente post, examinaremos las clases generales de reacciones en las que los complejos alquílicos son los principales actores, enfocándonos en los pasos mecanicistas específicos que involucran al complejo alquílico (eliminación reductora, transmetalación, inserción migratoria y [naturalmente] eliminación de β-hidruro).
Parte 3:
En este último post sobre ligandos alquílicos, exploraremos los principales modos de reactividad de los alquilos metálicos. Hemos discutido en detalle la eliminación de β-hidruro, pero otros destinos de los alquilos metálicos incluyen la eliminación reductiva, la transmetalación y la inserción migratoria en el enlace M-C. De manera similar a nuestros estudios de otros ligandos, nos gustaría relacionar las propiedades estéricas y electrónicas del complejo de alquilo metálico con su propensión a sufrir estas reacciones. Este tipo de pensamiento es particularmente importante cuando estamos interesados en controlar las tasas y/o grados relativos de dos vías de reacción diferentes y competidoras.
Reacciones de Complejos Metalquílicos
Recordemos que la eliminación de β-hidruro es una transformación extremadamente común, y a veces problemática, de alquilos metálicos. Por otra parte, hay reacciones para las que es deseable la eliminación de β-hidruro, como la reacción de Heck. Las modificaciones estructurales que fortalecen el enlace M—H en relación con el enlace M-C fomentan la eliminación de β-hidruro; la etapa también puede impulsarse atrapando el producto de hidruro metálico con una base (la reacción de Heck usa esta idea).
Por otro lado, la estabilización del enlace M-C desalienta la eliminación y fomenta su inversión: la inserción migratoria de olefinas en M-H. Anteriormente vimos el ejemplo de ligandos perfluoroalquilo, que poseen enlaces M-C excepcionalmente estables. La idea fundamental aquí, que los grupos aceptores de electrones en el ligando alquilo estabilizan el enlace M-C, es bastante general. Hartwig describe un aumento en el “carácter iónico” del enlace M-C tras la adición de grupos aceptores de electrones al ligando alquilo (fortaleciendo así el enlace M-C, ya que los enlaces iónicos son más fuertes que los enlaces covalentes). Las energías de enlace de la química orgánica confirman esta idea en cierta medida; por ejemplo, ver los BDE relativos de Me-Me, Me-Ph y Me-CCH en esta referencia. Todavía encuentro esta explicación un poco “ondulada a mano”, pero sirve a nuestro propósito, supongo.
Los alquilos metálicos están sujetos a eliminación reductora, el reverso microscópico de la adición oxidativa. El metal pierde dos ligandos covalentes, su estado de oxidación formal disminuye en dos unidades, el recuento total de electrones disminuye en dos unidades y se forma un enlace R—X. La eliminación reductiva es favorable cuando el enlace R-X en el producto orgánico es más estable que los enlaces M—R y M-X en el complejo inicial (una emisión termodinámica). Cabe señalar, sin embargo, que la cinética de eliminación reductora depende sustancialmente del volumen estérico de los ligandos eliminadores. La eliminación reductora concertada de R-H generalmente posee una energía de activación menor que la eliminación R-R.
La transmetalación implica la transferencia de un ligando alquílico de un metal al otro. Un problema interesante se refiere a la reactividad relativa de los alquilos metálicos hacia la transmetalación. Suponiendo conjuntos de ligandos similares y sin complicaciones, ¿cuál de los dos centros metálicos es más probable que se aferre a un ligando de alquilo? Considera las dos situaciones a continuación.
MR + M' M + M'R
MR + M'R' MR' + M'R
La primera es una transmetalación de buena fe; la segunda es realmente una reacción de doble reemplazo. La distinción rara vez se dibuja en la práctica, ¡pero es importante! La diferencia es que en el primer caso, se debe realizar una especie de transferencia de un solo electrón, mientras que en el segundo caso, no es necesaria una química redox. La favorabilidad en el primer caso se rige por los potenciales de reducción relativos de M y M' (la reacción avanza cuando M' se oxida más fácilmente que M); en el segundo caso, las electropositividades relativas de los metales son clave, y otros factores como las energías reticulares pueden ser importante. La distinción entre transmetalación per se y doble reemplazo explica la paradójica secuencia sintética en la figura siguiente. En la práctica, ambos se llaman “transmetalación”. Consulte estas diapositivas (página 6) para obtener una referencia resumida.
Esto lleva nuestra mirada extendida a los complejos de alquilo de metal a un cierre temporal. Por supuesto, los alquilos metálicos están en todas partes en química organometálica... ¡así que verlos de nuevo es prácticamente inevitable! La siguiente entrega de la serie Epic Ligand Survey se refiere a alilo, ciclopentadienilo y otros sistemas pi de miembros impares. Estos ligandos de tipo L n X pueden, como los arenos, apilar hasta seis electrones en el centro metálico a la vez.