
9.10: Espectros electrónicos de moléculas orgánicas
- Page ID
- 72810

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Características Generales
Un año después de que Herschel descubriera la radiación infrarroja, Johann Ritter descubrió la radiación más allá del extremo violeta del espectro visible. Esta radiación llegó a ser conocida como ultravioleta y pronto fue reconocida como especialmente efectiva en provocar reacciones químicas. La absorción de luz en las regiones ultravioleta y visible produce cambios en las energías electrónicas de las moléculas asociadas a la excitación de un electrón de un orbital estable a uno inestable. Debido a que la energía requerida para excitar los electrones de valencia-envoltura de las moléculas es comparable a las fuerzas de los enlaces químicos, la absorción puede conducir a reacciones químicas. Esto se discutió brevemente en el Capítulo 4 en relación con la halogenación fotoquímica de alcanos; en el Capítulo 28 se da un relato más detallado de la fotoquímica.
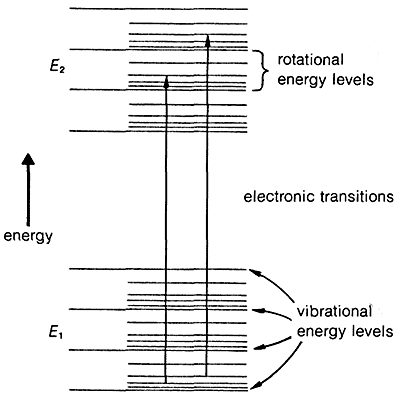
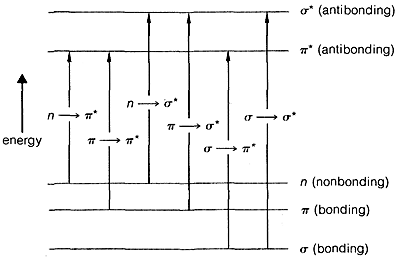
La transición de un electrón desde el estado fundamental\(E_1\), a un estado electrónico excitado\(E_2\), va acompañada de cambios vibracionales y rotacionales en la molécula, como se muestra en la Figura 9-17. En muestras de fase condensada, generalmente no es posible resolver las bandas de absorción resultantes lo suficientemente bien como para ver la estructura fina debido a las transiciones de vibración-rotación. En consecuencia, las absorciones debidas a la excitación electrónica son relativamente amplias.
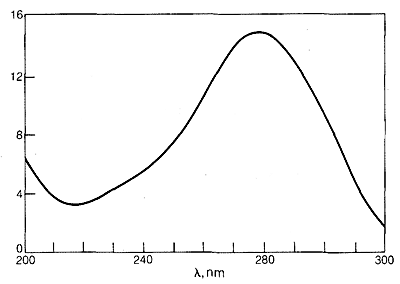
El espectro ultravioleta de la 2-propanona (acetona) se muestra en la Figura 9-18. La absorción débil, que alcanza los picos (es decir, tiene\(\lambda_\text{max}\) at\(280 \: \text{nm}\), es el resultado de la excitación de uno de los electrones no compartidos sobre el oxígeno a un nivel de energía más alto. Esto se llama transición\(n \rightarrow \pi^*\) (a menudo\(N \rightarrow A\)), en la que\(n\) denota que el electrón excitado es uno de los\(n\) electrones no compartidos en el oxígeno y\(\pi^*\) (estrella pi) denota que el electrón excitado va a un orbital antienlace de alta energía del carbono-oxígeno doble enlace (cf. Secciones 6-2 y 6-4C). El mismo tipo de\(n \rightarrow \pi^*\) transición ocurre aproximadamente a la misma longitud de onda e intensidad para muchos compuestos simples del tipo\(\ce{R_2C=O}\) y\(\ce{RCH=O}\), en el que\(R\) se encuentra un grupo alquilo. De una manera muy esquemática, podemos escribir
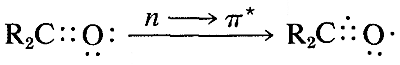
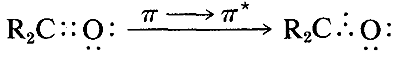

Efectos de la conjugación sobre espectros electrónicos
La\(\pi \rightarrow \pi^*\) transición para el eteno tiene\(\lambda_\text{max} = 175 \: \text{nm}\) y\(\epsilon_\text{max} = 10,000\). Se esperaría que un alcadieno daría un espectro de absorción similar al del eteno pero con un mayor\(\epsilon\), debido a que hay más dobles enlaces por mol para absorber la radiación. Esta expectativa se realiza más o menos para compuestos como 1,5-hexadieno y 1,3-dimetilenociclobutano, que tienen dobles enlaces aislados, pero no para 1,3-butadieno o etenilbenceno, que tienen dobles enlaces conjugados (Sección 3-3):
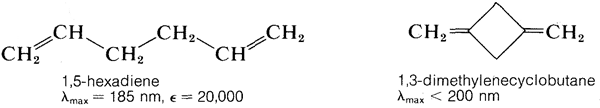

En general, los sistemas conjugados de dobles enlaces absorben radiación de longitudes de onda más largas y con mayor intensidad que los sistemas correspondientes de dobles enlaces aislados. Esto significa que la diferencia de energía entre los estados normal y excitado de los sistemas conjugados es menor que para los sistemas aislados de dobles enlaces. Para 1,3-butadieno y 1,5-hexadieno podemos calcular a partir de la Ecuación 9-2
que la energía de transición es aproximadamente\(23 \: \text{kcal}\) menor para el sistema conjugado. El estado fundamental del 1,3-butadieno se estabiliza quizás en\(3 \: \text{kcal}\) relación con un sistema no conjugado de dobles enlaces, lo que significa que el estado excitado debe estar mucho más estabilizado que esto si la energía de transición va a ser\(23 \: \text{kcal}\) menor que para 1,5-hexadieno.
¿Por qué el estado excitado de un sistema conjugado de dobles enlaces se estabiliza más, en relación con el estado fundamental, que para un sistema no conjugado? La teoría de resonancia proporciona una explicación (ver Sección 6-5). De las diversas estructuras convencionales de enlace de valencia que se pueden escribir para 1,3-butadieno, cuatro de las cuales se muestran aquí,\(2a\) -\(2d\), solo la estructura\(2a\) tiene una energía lo suficientemente baja como para ser dominante para el estado fundamental del 1,3-butadieno:
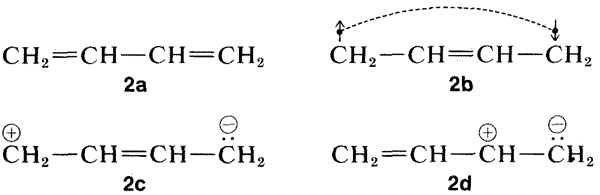
Ahora bien, cuando la molécula se excita en la medida de\(132 \: \text{kcal mol}^{-1}\) la luz\(217 \: \text{nm}\) ultravioleta, su energía es tan grande que esquemas de emparejamiento como\(2b\)\(2c\),\(2d\), y, que son demasiado desfavorables para contribuir mucho al estado fundamental, pueden ser muy importantes para el estado excitado. Así, se espera que la energía de estabilización del estado excitado, que tiene una multiplicidad de esquemas de emparejamiento de energía casi igual, sea mayor que la del estado fundamental con un esquema de emparejamiento dominante.
Cuantos más dobles enlaces haya en el sistema conjugado, menor será la diferencia de energía entre los estados normal y excitado. Los difenilpolienos de fórmula\(C_6H_5-(CH=CH)_n-C_6H_5\) absorben radiación a longitudes de onda progresivamente más largas a medida que\(n\) se incrementa. Esto es evidente a partir de los colores de los compuestos, que van desde incoloros con\(n = 1\)\(n = 2-7\), a naranja con\(n = 8\), a rojo con, como\(\lambda_\text{max}\) va del ultravioleta a la región visible del espectro electromagnético.
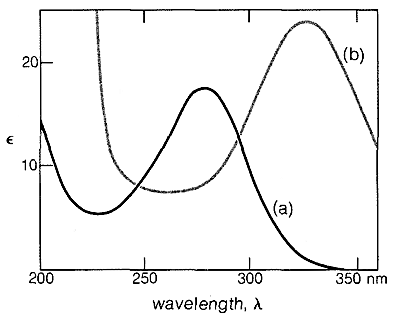
Efectos similares se encuentran con enlaces conjugados\(\ce{C=O}\) y\(\ce{C=N}\) dobles. Por ejemplo, los espectros electrónicos de 2-butanona y 3-buten-2-ona se muestran en la Figura 9-20.
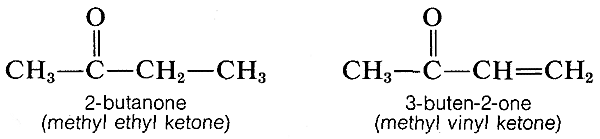
La conjugación también puede influir en espectros infrarrojos. Las transiciones que surgen de\(C=C\) y las vibraciones de\(C=O\) estiramiento generalmente son más intensas y se desplazan a frecuencias ligeramente más bajas (longitudes de onda más largas) para compuestos conjugados en relación con compuestos no conjugados. Así el\(C=C\) estiramiento de 1-buteno ocurre en\(1650 \: \text{cm}^{-1}\), mientras que el de 1,3-butadieno se observa en\(1597 \: \text{cm}^{-1}\).
Los alcanos y cicloalcanos no tienen transiciones electrónicas de baja energía comparables a los sistemas conjugados o moléculas con electrones no enlazantes. Por lo tanto, los alcanos y cicloalcanos no muestran absorción por encima\(200 \: \text{nm}\) y son buenos solventes para usar en espectroscopía electrónica.
Aplicaciones de la Espectroscopia Electrónica
¿Cómo utilizamos la espectroscopia electrónica en el análisis químico? Las dos aplicaciones principales son determinaciones de estructura y análisis cuantitativo. La posición e intensidad de una banda de absorción electrónica proporciona información sobre la estructura química. Tales absorciones normalmente no son tan útiles como las absorciones infrarrojas porque no dan como información detallada. Para nuestros propósitos aquí, los principales puntos a recordar son:
- Una absorción débil (\(\epsilon = 10-100\)) sugiere una\(n \rightarrow \pi^*\) transición de un grupo carbonilo aislado. Si esta absorción se encuentra en la región\(270\) - es probable\(350 \: \text{nm}\) un aldehído o cetona.
- Absorciones algo más fuertes (\(\epsilon = 100-4,000\)) entre\(200 \: \text{nm}\) y\(260 \: \text{nm}\) pueden corresponder a\(\pi \rightarrow \sigma^*\) transiciones.
- Las fuertes absorciones (\(\epsilon = 10,000-20,000\)) suelen ser características de\(\pi \rightarrow \pi^*\) las transiciones. Si la absorción ocurre arriba\(200 \: \text{nm}\), se indica un sistema conjugado de múltiples enlaces. Cada doble enlace carbono-carbono adicional se desplaza\(\lambda_\text{max}\) aproximadamente\(30 \: \text{nm}\) a longitudes de onda más largas y mejora la intensidad de absorción. La conjugación también cambia\(\lambda_\text{max}\) de\(n \rightarrow \pi^*\) transiciones a longitudes de onda más largas.
Si se trata de compuestos para los que se conocen las longitudes de onda y las intensidades molares de las bandas de absorción, entonces podemos usar el grado de absorción para el análisis cuantitativo con la ayuda de la ley Beer-Lambert (ver Tabla 9-3 para definiciones):
\[A = \epsilon cl\]
Al medir la absorbancia\(A\) de una muestra conocida\(\epsilon\) en una celda de longitud de trayectoria conocida\(I\), se\(c\) puede determinar la concentración. Debido a que los cambios en la absorbancia reflejan cambios en la concentración, es posible usar mediciones de absorbancia para seguir las tasas de reacciones químicas, para determinar las constantes de equilibrio (como las constantes de disociación de ácidos y bases) y seguir cambios conformacionales en moléculas bioorgánicas como proteínas y ácidos nucleicos.
Se ha realizado la capacidad de la espectroscopia electrónica para realizar cualitativa y cuantitativa de los elementos en compuestos químicos y se han realizado aplicaciones a elementos de bajo número atómico, como el carbono y el oxígeno. Los espectros electrónicos se han desarrollado en el estudio de estos elementos y sus compuestos.
Referencias
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."