
9.11: Espectroscopia de Resonancia Magnética Nuclear
- Page ID
- 72843

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)La espectroscopia de resonancia magnética nuclear (RMN) es extremadamente útil para la identificación y análisis de compuestos orgánicos. El principio en el que se basa esta forma de espectroscopia es simple. Los núcleos de muchos tipos de átomos actúan como pequeños imanes y tienden a alinearse en un campo magnético. En espectroscopía RMN, medimos la energía requerida para cambiar la alineación de núcleos magnéticos en un campo magnético. Para ilustrar el procedimiento con un ejemplo sencillo, considere el comportamiento de un protón\(\left( ^1H \right)\) en un campo magnético. Existen dos posibles alineaciones de este núcleo magnético con respecto a la dirección del campo aplicado, como se muestra en la Figura 9-21. Los imanes nucleares pueden estar alineados ya sea con la dirección del campo, u opuestos a ella. Las dos orientaciones no son equivalentes, y se requiere energía para cambiar la alineación más estable a la alineación menos estable.

En la Figura 9-22 se muestra un diagrama esquemático de un instrumento de RMN. Cuando una sustancia como el etanol\(CH_3-CH_2-OH\), cuyos hidrógenos tienen núcleos (protones) que son magnéticos, se coloca en la bobina transmisora y el campo magnético se incrementa gradualmente, a ciertas intensidades de campo la energía de radiofrecuencia es absorbida por la muestra y el amperímetro indica un aumento en el flujo de corriente en la bobina. El resultado global es un espectro como el que se muestra en la Figura 9-23. Este espectro es lo suficientemente detallado como para servir como una “huella digital” útil para el etanol, y además es lo suficientemente simple como para que podamos dar cuenta del origen de cada línea. El propósito de esta sección es explicar cómo las complejidades de espectros como el de la Figura 9-23 pueden interpretarse en términos de estructura química.

¿Para qué tipo de sustancias podemos esperar que ocurra la absorción por resonancia magnética nuclear? Las propiedades magnéticas siempre se encuentran con núcleos de masas impares,\(^1H\),\(^{13}C\),\(^{15}N\),\(^{17}O\),\(^{19}F\),\(^{31}P\), y así sucesivamente, así como para núcleos de masa par pero número atómico impar\(^2H\),\(^{10}B\),\(^{14}N\), y así sucesivamente. \(^8\)Núcleos como\(^{12}C\),\(^{16}O\), y\(^{32}S\), que tienen incluso números másicos y atómicos, no tienen propiedades magnéticas y no dan señales de resonancia magnética nuclear. Por diversas razones, el uso rutinario de espectros de RMN en química orgánica se limita a\(^1H\)\(^{19}F\),,\(^{13}C\), y\(^{31}P\). En este capítulo nos ocuparemos únicamente de los espectros de RMN de hidrógeno (\(^1H\)) y de carbono (\(^{13}C\)).

El tipo de espectroscopía de RMN que discutiremos aquí es limitado en sus aplicaciones ya que solo se puede realizar con líquidos o soluciones. Afortunadamente, el rango permisible de solventes es grande, desde hidrocarburos hasta ácido sulfúrico concentrado, y para la mayoría de los compuestos es posible encontrar un solvente adecuado.
Los espectros de resonancia magnética nuclear pueden ser tan simples como para tener un solo pico de absorción, pero también pueden ser mucho más complejos que el espectro de la Figura 9-23. Sin embargo, es importante reconocer que no importa cuán complejo parezca ser un espectro de RMN, implica solo tres parámetros: desplazamientos químicos, divisiones espín-espín y procesos cinéticos (tasa de reacción). Tendremos más que decir sobre cada uno de estos más adelante. Primero, intentemos establecer la relación de la espectroscopia de RMN con algunas de las otras formas de espectroscopia que ya hemos discutido en este capítulo.
La relación de RMN con otros tipos de espectroscopía
La\(^9\) espectroscopia de resonancia magnética nuclear implica transiciones entre posibles niveles de energía de núcleos magnéticos en un campo magnético aplicado (ver Figura 9-21). Las energías de transición están relacionadas con la frecuencia de la radiación absorbida por la ecuación familiar\(\Delta E - h \nu\). Una diferencia importante entre la RMN y otras formas de espectroscopia es que\(\Delta E\) está influenciada por la fuerza del campo aplicado. Esto no debería ser sorprendente, porque si vamos a medir la energía de cambiar la dirección de alineación de un núcleo magnético en un campo magnético, entonces cuanto más fuerte sea el campo, más energía será invamada.
El espín nuclear (simbolizado como\(I\)) es una propiedad cuantificada que se correlaciona con el magnetismo nuclear de tal manera que cuando\(I\) es cero el núcleo no tiene espín ni propiedades magnéticas. Los ejemplos son\(^{12}C\) y\(^{16}O\). Varios núcleos de particular interés para los químicos orgánicos -\(^1H\),\(^{13}C\),\(^{15}\)\(^{19}\), y\(^{31}P\) - tienen giro de\(\frac{1}{2}\). Con sólo\(I = \frac{1}{2}\) hay dos estados de energía magnética del núcleo en un campo magnético. Estos estados se designan con los números cuánticos de espín\(+ \frac{1}{2}\) y\(- \frac{1}{2}\). La diferencia de energía entre estos estados,\(\Delta E\), viene dada por
\[\Delta E = \gamma h H = h \nu\]
o
\[\nu = \gamma H\]
en la que\(h\) es la constante de Planck,\(\nu\) está en hercios,\(\gamma\) es una constante magnética nuclear llamada relación giromagnética,\(^{10}\), y\(H\) es la intensidad del campo magnético en el núcleo. En general, no\(H\) será exactamente igual a\(H_\text{o}\), el campo magnético aplicado y, como veremos, esta diferencia lleva a información química importante. Cada tipo de núcleo (\(^1H\),\(^{13}C\),\(^{15}N\), etc.) tiene su propio\(\gamma\) valor y, en consecuencia, sufrirá transiciones a diferentes frecuencias a cualquier valor particular de\(H\). Esto debería ser más claro mediante el estudio de la Figura 9-24.

\ (^1H\) y\(^{13}C\) núcleos en función del campo magnético. La escala vertical es de frecuencia\(\nu\) en\(\text{MHz}\) (\(= 10^4 \: \text{Hz} = 10^6\)ciclos de 1 megahercios por segundo) mientras que la escala horizontal es de campo magnético en gauss. (A modo de comparación, el campo magnético de la Tierra es de aproximadamente 0.2 gauss.) La línea vertical discontinua en\(14,100\) gauss nos dice que la frecuencia de\(^1H\) resonancia será\(60.0 \: \text{MHz}\) y la frecuencia de\(^{13}C\) resonancia estará\(15.0 \: \text{MHz}\) en esta intensidad de campo. </em">
Existen varios modos de funcionamiento de un espectrómetro de RMN. Primero y más común, mantenemos\(\nu\) constantes y variamos (o “barrer”)\(H_\text{o}\). Cerca de\(\nu = \gamma H\), la energía es absorbida por los núcleos y el flujo de corriente desde el transmisor aumenta hasta que\(\nu\) es exactamente igual a\(\gamma H\). Incremento adicional de\(H_\text{o}\) marcas\(\nu < \gamma H_\text{o}\) y disminuye el flujo de corriente. La forma de la curva de absorción de energía en función de\(H_\text{o}\) cuándo\(H_\text{o}\) se cambia muy lentamente se muestra en la Figura 9-25a. El pico se centra en el punto donde\(\nu = \gamma H\). Cuando\(H_\text{o}\) se cambia más rápidamente, se observan efectos transitorios en el pico, los cuales son consecuencia de que los núcleos no revierten instantáneamente del\(+ \frac{1}{2}\) estado\(- \frac{1}{2}\) a. El resultante
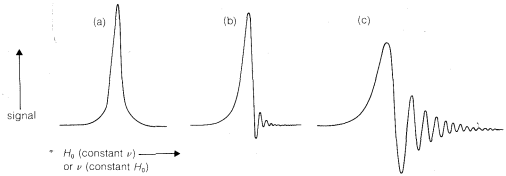
fenómeno se llama “timbre” y se muestra en las Figuras 9-25b y 9-25c. También se observará evidencia de zumbido en los picos de la Figura 9-23.
Un método alternativo para hacer funcionar un espectrómetro de RMN es mantener constante el campo magnético y barrer la frecuencia del transmisor a través de las resonancias. Este modo de operación se parece más a otras formas de espectroscopia y da las mismas formas de línea que barrer el campo (Figura 9-25).
¿Qué energía está asociada con una transición de\(^1H\) RMN? La magnitud de esta energía puede calcularse a partir de la relación entre energía y longitud de onda (frecuencia) de la radiación absorbida (Sección 9-4). Es decir,
\[\Delta E = \frac{28,600}{\lambda} \text{kcal mol}^{-1}\]y\[\lambda = \frac{c}{\nu}\]
La frecuencia\(\nu\) es la frecuencia de funcionamiento del espectrómetro, que tomaremos como\(60 \: \text{MHz}\) o\(6 \times 10^7 \: \text{Hz}\) (ciclos\(\text{sec}^{-1}\)), y la velocidad de la luz es\(3 \times 10^8 \: \text{m sec}^{-1}\). De ahí
\[\lambda = \frac{3 \times 10^8 \times 10^9 \left( \text{nm sec}^{-1} \right)}{6 \times 10^7 \left( \text{Hz} \right)} = 5 \times 10^9 \: \text{nm}\]
y
\[\Delta E = \frac{28,600}{5 \times 10^9} = 5.7 \times 10^{-6} \: \text{kcal mol}^{-1}\]
Esta es una diferencia de energía muy pequeña, lo que significa que solo muy pocos núcleos más están en el\(+ \frac{1}{2}\) estado más estable que en el\(- \frac{1}{2}\) estado menos estable. ¡La constante de equilibrio\(K\) para\(- \frac{1}{2} \rightleftharpoons + \frac{1}{2}\) calculada a partir de la Ecuación 4-2 para\(25^\text{o}\) (298\:\ text {K}\)) y descuidar posibles efectos de entropía es de 1.000010!
El cambio químico
La gráfica de señal contra la intensidad del campo magnético para etanol en la Figura 9-23 muestra tres grupos principales de líneas correspondientes a las tres variedades de hidrógeno presentes: metilo (\(CH_3\)), metileno (\(CH_3\)) e hidroxilo (\(OH\)). Las diferencias en las intensidades de campo a las que se obtienen señales para núcleos del mismo tipo, como los protones, pero ubicados en diferentes ambientes moleculares, se denominan desplazamientos químicos.
Otro punto muy importante a notar sobre la Figura 9-23 es que las intensidades de las tres absorciones principales están en la relación de 1:2:3, correspondiente a la relación del número de cada tipo de protón (\(OH\),\(CH_2\),\(CH_3\)) que produce la señal. En general, las áreas bajo los picos de un espectro como en la Figura 9-23 son proporcionales al número de núcleos en la muestra que dan esos picos. Las áreas se pueden medir por integración electrónica y la integral a menudo se muestra en el gráfico, como lo está en la Figura 9-23, como una línea escalonada que aumenta de izquierda a derecha. La altura de cada escalón corresponde al número relativo de núcleos de un tipo particular. A menos que se tomen precauciones especiales, las integrales generalmente no deben considerarse precisas a mejores que aproximadamente\(5\%\).
¿Por qué los protones en diferentes ambientes moleculares se absorben a diferentes intensidades de campo? La intensidad del campo\(H\) en un núcleo particular es menor que la intensidad del campo magnético externo\(H_\text{o}\). Esto se debe a que los electrones de valencia alrededor de un núcleo particular y alrededor de núcleos vecinos responden al campo magnético aplicado para proteger al núcleo del campo aplicado. La forma en que ocurre este blindaje es la siguiente.
Primero, cuando un átomo es colocado en un campo magnético, sus electrones son forzados a someterse a una rotación alrededor del eje del campo, como se muestra en la Figura 9-26. Segundo,
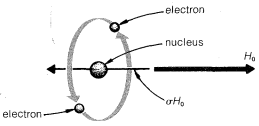
rotación de los electrones alrededor del núcleo es una circulación de carga, y esto crea un pequeño campo magnético en el núcleo opuesto en la dirección a\(H_\text{o}\). Tercero, la magnitud de este\(^{11}\) efecto diamagnético es directamente proporcional\(H_\text{o}\) y se puede cuantificar como\(\sigma H_\text{o}\), en la que\(\sigma\) se encuentra la constante de proporcionalidad. Es importante reconocer que no\(\sigma\) es una propiedad nuclear sino que depende del ambiente químico del átomo. Cada protón químicamente diferente tendrá un valor diferente\(\sigma\) y, por lo tanto, un cambio químico diferente.
El campo real\(H\) en el núcleo será\(H_\text{o} - \sigma H_\text{o}\). Debido a que\(\sigma\) actúa para reducir la intensidad del campo aplicado en el núcleo, se le llama el parámetro de blindaje magnético. Cuanto más blindaje haya, más fuerte debe ser el campo aplicado para satisfacer la condición de resonancia,
El uso común es: campo arriba, más blindaje; campo abajo, menos blindaje; y debe recordar que los espectros de barrido de campo siempre se registran con el campo aumentando de izquierda a derecha.

Desplazamiento Químico y Estereoquímica
El valor de la espectroscopia de RMN en la determinación de la estructura radica en el hecho de que los núcleos químicamente diferentes absorben a diferentes intensidades de campo. En secciones posteriores nos ocuparemos de correlacionar los cambios químicos con las características estructurales. No obstante, antes de continuar adelante es sumamente importante que sea capaz de identificar el número y tipo de protones no equivalentes en una estructura dada, y por lo tanto el número de desplazamientos químicos a esperar. Este número no siempre es evidente por sí mismo, especialmente cuando intervienen sutiles factores de estereoquímica. Por esta razón, le sugerimos que inspeccione las estructuras\(3\),\(5\) para convencerse de que los protones etiquetados con diferentes subíndices de letras en cualquier molécula son en efecto químicamente diferentes.
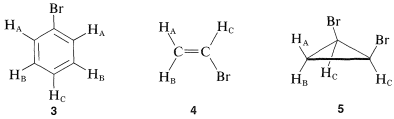
Una forma de verificar si dos protones están en entornos equivalentes es imaginar que cada uno es reemplazado por separado por un átomo o grupo diferente. Si el producto de reemplazar\(H_\text{A}\) es idéntico al obtenido al reemplazar\(H_\text{B}\), entonces\(H_\text{A}\) y\(H_\text{B}\) son químicamente equivalentes. Si los dos productos son no idénticos, entonces\(H_\text{A}\) y no\(H_\text{B}\) son equivalentes. Por ejemplo, la sustitución de\(H_\text{A}\) o\(H_\text{B}\) en\(3\)\(4\), y\(5\) por un átomo\(X\) daría diferentes productos. Por lo tanto,\(H_\text{A}\) y no\(H_\text{B}\) son equivalentes en\(3\),\(4\), y\(5\).
Las cosas se complican más con sustancias como\(6\) y\(7\):

Observe que\(6\) representa una molécula quiral y si\(H_\text{A}\) y\(H_\text{B}\) cada uno se reemplazan con\(X\) obtenemos\(8\) y\(9\), que son diastereómeros (ver Sección 5-5). Se puede verificar esto con modelos moleculares si es necesario. Los diastereómeros tienen diferentes propiedades químicas y físicas; por lo tanto\(H_\text{A}\) y\(H_\text{B}\) en no\(6\) son equivalentes. A menudo se les llama hidrógenos diastereotópicos.

¿Qué hay de los dos protones de metileno en etanol\(7\),, que hemos etiquetado como\(H_\text{A}\)\(H_\text{A'}\)? ¿Son idénticos? En cierto sentido no son idénticos porque, si cada uno fuera reemplazado por\(X\), tendríamos un par de enantiómeros. Por lo tanto,\(H_\text{A}\) y en\(H_\text{A'}\) ocasiones se llaman hidrógenos enantiotópicos.

Pero, recordarán que los enantiómeros son químicamente indistinguibles a menos que estén en un ambiente quiral. Por lo tanto, esperamos que los cambios de los hidrógenos enantiotópicos sean idénticos, a menos que estén en un ambiente quiral. En resumen, los protones enantiotópicos normalmente tendrán los mismos desplazamientos químicos, mientras que los protones diastereotópicos normalmente tendrán diferentes desplazamientos químicos.
Hasta ahora hemos ignorado la relación de los cambios químicos con los equilibrios conformacionales. Considere un ejemplo específico, 1,2-dibromoetano, para el cual hay tres conformaciones escalonadas\(10a\)\(10b\), y\(10c\):
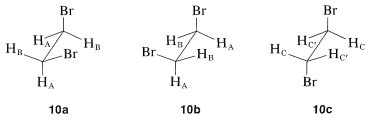
Se espera que cada una de estas conformaciones tenga su propio espectro de RMN. Las dos formas gauche,\(10a\) y\(10b\), son enantiómeros y sus espectros deben ser idénticos. Los hidrógenos\(H_\text{A}\) en\(10a\) cada uno son trans al bromo en el carbono adyacente, mientras que los\(H_\text{B}\) hidrógenos son cis a los mismos bromo (ver Sección 5-5A). En consecuencia, los\(H_\text{B}\) hidrógenos\(H_\text{A}\) y los hidrógenos no son equivalentes y se esperaría que tuvieran diferentes cambios químicos. En contraste, todos los hidrógenos del anticonfórmero,\(10c\), son equivalentes y tendrían el mismo desplazamiento químico. Por lo tanto, esperaríamos observar tres cambios químicos derivados de\(H_\text{A}\),\(H_\text{B}\), y\(H_\text{C}\) para una mezcla de\(10a\),\(10b\), y\(10c\). Sin embargo, el espectro real de 1,2-dibromoetano muestra solo una señal aguda de protones en condiciones ordinarias. La razón es que los núcleos magnéticos pueden absorber la radiación excitante. El resultado es que observamos un desplazamiento químico promedio, que refleja los cambios relativos y poblaciones de los tres confórmeros presentes. Si podemos ir a una temperatura suficientemente baja como para hacer lenta la interconversión de las conformaciones (del orden de 10 veces por segundo), entonces esperaremos ver los tres cambios químicos diferentes\(H_\text{A}\),\(H_\text{B}\), y\(H_\text{C}\) con intensidades correspondientes a las poblaciones reales del conformaciones a la temperatura de la muestra. Este es un ejemplo del efecto de los procesos de velocidad sobre los espectros de RMN. Otros ejemplos y un relato más detallado de cómo relacionar la apariencia de la señal con los tipos de cambio de los procesos cambiarios se dan en la Sección 27-2.
Estándares y unidades de turno químico
Los desplazamientos químicos siempre se miden con referencia a un estándar. Para los protones o\(^{13}C\) en las moléculas orgánicas, el estándar habitual es un tetrametilsilano\(\left( CH_3 \right)_4 Si\), que da señales de RMN fuertes y nítidas en regiones donde solo unos pocos otros tipos de protones o núcleos de carbono absorben. Los desplazamientos químicos a menudo se expresan en\(\text{Hz}\) (ciclos por segundo) en relación con el tetrametilsilano (TMS). Estas pueden parecer unidades extrañas para la intensidad del campo magnético, pero debido a que la resonancia ocurre en\(\nu = \gamma H\), ya sea unidades de frecuencia (\(\text{Hz}\), radianes\(\text{sec}^{-1}\)) o unidades de campo magnético (gauss) son apropiadas.
Hace diez años, la mayoría de los espectrómetros de RMN operaban para protones con transmisores de radiofrecuencia\(60 \: \text{MHz}\) (rf) establecidos en (\(6 \times 10^7\)ciclos por segundo) pero ha habido una proliferación de diferentes frecuencias de operación de protones y ahora\(30\)\(60\),\(90\),\(100\),\(220\),\(270\), \(300\)y\(360 \: \text{MHz}\) las máquinas están disponibles comercialmente. El costo de estas máquinas es aproximadamente proporcional al cuadrado de la frecuencia, y uno bien puede preguntarse por qué hay disponible una variedad tan exótica y qué tiene que ver esto con el cambio químico. Las altas frecuencias de operación son deseables porque los cambios químicos aumentan con la frecuencia del espectrómetro, y esto hace que los espectros sean más simples de interpretar Un aumento de 12 veces en la frecuencia de operación (como de\(30 \: \text{MHz}\) a\(360 \: \text{MHz}\)) significa un aumento de 12 veces\(H_\text{o}\) en el punto de resonancia (recuerde\(\nu = \gamma H\)) y esto significa también un aumento de 12 veces en\(\sigma H_\text{o}\). Por lo tanto, las resonancias que difieren porque corresponden a diferentes\(\sigma\) valores estarán doce veces más separadas en\(360 \: \text{MHz}\) que en\(30 \: \text{MHz}\). Esto puede producir una dramática simplificación de los espectros, como puede verse en la Figura 9-27, que muestra el efecto de casi un factor de cuatro\(\nu\) en el espectro de RMN protónica del 2-metil-2-butanol. \(^{12}\)
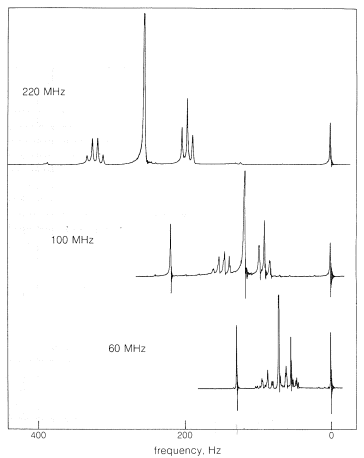
Para reiterar, los desplazamientos químicos son estrictamente proporcionales a la frecuencia del espectrómetro, por lo que las líneas\(100 \: \text{Hz}\) separadas a\(60 \: \text{MHz}\) estarán\(167 \: \text{Hz}\) separadas en\(100 \: \text{MHz}\). Esto podría parecer hacer comparaciones de espectros de RMN en diferentes espectrómetros irremediablemente complejas pero, debido a la proporcionalidad de los desplazamientos a frecuencia (o campo), si dividimos los cambios medidos en\(\text{Hz}\) (relativos al mismo estándar) para cualquier espectrómetro por el transmisor frecuencia en\(\text{MHz}\), obtenemos un conjunto de cambios independientes de la frecuencia en partes por millón (\(\text{ppm}\), que son útiles para todos los espectrómetros de RMN. Los desplazamientos de RMN reportados en\(\text{ppm}\) relación con TMS como cero, como se muestra en la Figura 9-23, se denominan valores\(\delta\) (delta):
\[\delta = \frac{\left( \text{chemical shift downfield in Hz relative to TMS} \right) \times 10^6}{\text{spectrometer frequency in Hz}}\]
Por lo tanto, si a\(60 \: \text{MHz}\) una señal de protón viene\(100 \: \text{Hz}\) campo abajo con respecto al tetrametilsilano, se puede designar como\(\left( +100 \: \text{Hz} \times 10^6 \right)/ 60 \times 10^6 \: \text{Hz} = +1.67 : \text{ppm}\) relativa al tetrametilsilano. En\(100 \: \text{MHz}\), la línea entonces será\(\left( 1.67 \times 10^{-6} \right) \left(100 \times 10^6 \right) = 167 \: \text{Hz}\) campo abajo de tetrametilsilano. Los desplazamientos químicos típicos de protones en relación con el TMS se dan en la Tabla 9-4. \(^{13}\)Los valores citados para cada tipo de protón pueden, en la práctica, mostrar variaciones de\(0.1\) -\(0.3 \: \text{ppm}\). Esto no es irrazonable, porque se espera que el desplazamiento químico de un protón determinado dependa algo de la naturaleza de la molécula en particular involucrada, y también del disolvente, la temperatura y la concentración.
Un\(\delta\) valor positivo significa un desplazamiento a un campo más bajo (o menor frecuencia) con respecto a TMS, mientras que un negativo\(\delta\) significa un desplazamiento a un campo más alto (o mayor frecuencia). La\(\delta\) convención es ampliamente aceptada, pero a menudo se encuentran en la literatura cambios de protones con referencia a TMS reportados como "\(\tau\)valores”. La\(\tau\) escala tiene la referencia TMS en\(+10 \: \text{ppm}\), por lo que la mayoría de las señales de protones caen en el rango de\(\tau = 0\) a\(\tau = +10\). Un\(\tau\) valor se puede convertir al\(\delta\) valor apropiado restándolo de 10. La vida con espectros de RMN sería más sencilla si la\(\tau\) escala simplemente desaparecería.
Correlaciones entre la estructura y los cambios químicos
Los cambios químicos protónicos son muy valiosos para la determinación de estructuras, pero para utilizar los cambios de esta manera debemos saber algo sobre las correlaciones que existen entre el desplazamiento químico y el ambiente estructural de los protones en los compuestos orgánicos. Los efectos más importantes surgen de las diferencias en la electronegatividad, los tipos de enlaces de carbono, los enlaces de hidrógeno y el intercambio químico.
Electronegatividad
Considere primero los desplazamientos químicos de los protones adheridos a un\(sp^3\) carbono, .
.
El grado de blindaje del protón por los electrones de valencia de carbono depende del carácter de los átomos y grupos sustituyentes presentes, y particularmente de su poder de atracción de electrones, o electronegatividad. Para una agrupación del tipo , el blindaje será menor ya que\(\ce{X}\) es más extracción de electrones en relación con el hidrógeno:
, el blindaje será menor ya que\(\ce{X}\) es más extracción de electrones en relación con el hidrógeno:
 Si\(\ce{X}\) es electrón-retirando, el protón es deshidado.
Si\(\ce{X}\) es electrón-retirando, el protón es deshidado.
Por ejemplo, los desplazamientos químicos de protones de los haluros de metilo (Cuadro 9-4) muestran un blindaje decreciente, por lo tanto, desplazamientos químicos de campo bajo progresivamente al aumentar la electronegatividad del halógeno\(\left( \ce{F} > \ce{Cl} > \ce{Br} > \ce{I} \right)\):

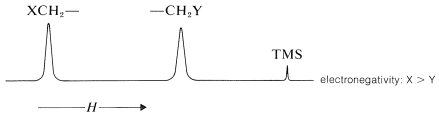
Cuadro 9-4: Valores típicos de desplazamiento químico de protones\(\left( \delta \right)\) en\(\ce{CHCl_3}\) soluciones diluidas


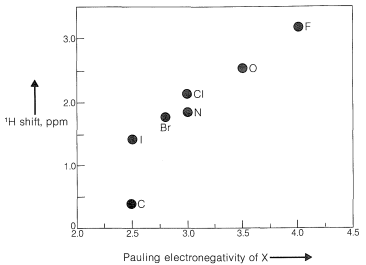
Cuando dos grupos electronegativos,\(\ce{X}\) y\(\ce{Y}\), están unidos al mismo carbono, como en\(\ce{XCH_2Y}\), se espera que los protones estén menos protegidos y entren en resonancia campo abajo de los metilenos de\(\ce{XCH_2CH_2Y}\). Existe una relación aproximada (ver abajo) entre los desplazamientos de los\(\ce{XCH_2Y}\) protones y las constantes\(\left( \sigma \right)\) de blindaje efectivas de\(\ce{X}\) y\(\ce{Y}\) conocidas como regla de Shoolery.
\[\delta = 0.23 + \sigma_x + \sigma_y \tag{9-4}\]
Los valores apropiados de\(\sigma\) para su uso con esta ecuación se dan en la Tabla 9-4.
Efectos del tipo de enlace de carbono
Los desplazamientos de los protones de alcanos y cicloalcanos caen en el rango de\(0.9\) -\(1.5 \: \text{ppm}\) con\(\ce{C-H}\) protones llegando al extremo de campo bajo de este rango y\(\ce{-CH_3}\) protones llegando al extremo de campo alto (ver Cuadro 9-4).
Los hidrógenos alquénicos (hidrógenos vinílicos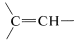 ) normalmente se observan entre\(4.6\) -\(6.3 \: \text{ppm}\) hacia campos más bajos que los desplazamientos de protones en alcanos y cicloalcanos. Esto significa que los hidrógenos alquénicos en un compuesto orgánico pueden distinguirse fácilmente de los hidrógenos alcanos.
) normalmente se observan entre\(4.6\) -\(6.3 \: \text{ppm}\) hacia campos más bajos que los desplazamientos de protones en alcanos y cicloalcanos. Esto significa que los hidrógenos alquénicos en un compuesto orgánico pueden distinguirse fácilmente de los hidrógenos alcanos.
Los protones aromáticos, como los del benceno, tienen desplazamientos en campos aún más bajos y comúnmente se observan en\(7\) -\(8 \: \text{ppm}\). En contraste, los protones alquínicos del tipo\(\ce{-C \equiv CH}\) dan resonancias que son campo alto de protones alquénicos o aromáticos y llegan a\(2\) -\(3 \: \text{ppm}\). Otro efecto asociado con múltiples enlaces es la gran diferencia en el desplazamiento entre un\(\ce{-CH(OCH_3)_2}\) protón, que normalmente llega a aproximadamente\(5.5 \: \text{ppm}\), y los protones de aldehído,\(\ce{-CH=O}\), que son mucho más pedos campo abajo en\(9\) -\(11 \: \text{ppm}\).
Claramente, los desplazamientos de un protón dependen de si el carbono forma enlaces simples, dobles o triples. En un campo magnético, la circulación de electrones en los\(\pi\) orbitales de múltiples enlaces inducidos por el campo (Figura 9-26) genera efectos de blindaje diamagnético en algunas regiones del enlace múltiple y efectos de deshielding paramagnéticos en otras regiones. Al parecer, los protones unidos a los carbonos de doble enlace se encuentran en las zonas de desblindaje y, por lo tanto, están en campo abajo, mientras que los protones unidos a los carbonos de triple enlace están en las zonas de blindaje y se observan en un campo bastante alto
Enlace de hidrógeno
Cuando un protón está unido directamente a un átomo fuertemente electronegativo como el oxígeno o el nitrógeno, su desplazamiento químico depende críticamente de la naturaleza del disolvente, la temperatura, la concentración y si están presentes impurezas ácidas o básicas. Las variaciones habituales en el desplazamiento químico para tales protones son tan grandes (hasta\(5 \: \text{ppm}\) para los alcoholes) que no existen correlaciones muy útiles.
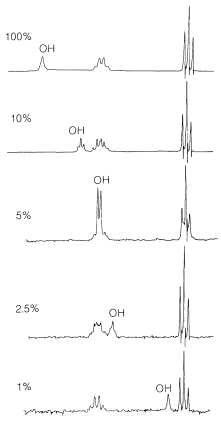
Los enlaces de hidrógeno son la razón principal de los desplazamientos químicos variables de\(\ce{OH}\) los\(\ce{NH}\) protones. En general, los enlaces de hidrógeno resultan en deshielding, lo que hace que las resonancias se muevan campo abajo. La extensión de los enlaces de hidrógeno varía con la concentración, la temperatura y el disolvente, y los cambios en el grado de enlace de hidrógeno pueden causar cambios sustanciales de desplazamiento. Esto es muy evidente en el espectro de RMN del etanol tomado a diferentes concentraciones en\(\ce{CCl_4}\) (Figura 9-29). Se verá que la resonancia de hidroxilo se mueve campo arriba mediante enlaces de hidrógeno a través de equilibrios como
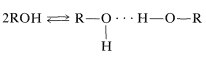
Intercambio Químico
Muchos\(\ce{OH}\) y\(\ce{NH}\) compuestos son ácidos débiles y bases débiles y pueden sufrir autoprotólisis, lo que significa que un protón puede transferirse de una molécula a otra. Supongamos que tenemos un compuesto como el 2-aminoetanol,\(\ce{H_2NCH_2CH_2OH}\). Normalmente se esperaría que esta sustancia tuviera una resonancia de\(\ce{NH_2}\) protones aproximadamente\(1 \: \text{ppm}\) y una resonancia de\(\ce{OH}\) protones aproximadamente\(3 \: \text{ppm}\). Los equilibrios de autoprotólisis pueden intercambiar los protones entre las moléculas y también de un extremo a otro como se muestra a continuación, aunque los equilibrios no sean muy favorables.
\[\begin{align} \ce{NH_2CH_2CH_2OH} &\overset{\rightarrow}{\longleftarrow} ^\oplus \ce{NH_3CH_2CH_2O}^\ominus \tag{9-5} \\ 2 \ce{NH_2CH_2CH_2OH} &\overset{\rightarrow}{\longleftarrow} ^\oplus \ce{NH_3CH_2CH_2OH} + \ce{N_2CH_2CH_2O}^\ominus \tag{9-6} \end{align}\]
Dichos equilibrios se pueden establecer muy rápidamente, especialmente si están presentes trazas de un ácido fuerte o una base fuerte. En tales circunstancias, se observa una única señal de\(\left( \ce{-NH_2}, \: \ce{-OH} \right)\) protón promedio, debido a que la excitación de un protón dado de su estado magnético de menor energía a su estado magnético de mayor energía ocurre mientras está parcialmente en oxígeno y en parte en nitrógeno. Este es el mismo tipo de promedio de desplazamiento químico que ocurre para las conformaciones que equilibran rápidamente (ver Sección 9-10C).
Aplicación de Cambios Químicos a la Determinación de Estructura
Para ver cómo se pueden usar juntos los espectros de RMN e infrarrojos para la determinación de la estructura, trabajaremos a través de un ejemplo representativo.
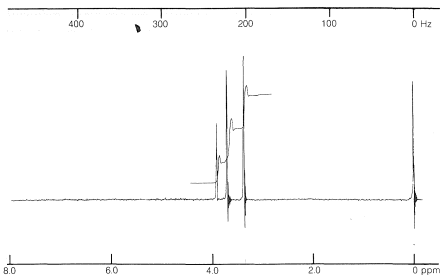
El objetivo es asignar una estructura al compuesto\(\ce{C_4H_8O_3}\) cuyo espectro de RMN se muestra en la Figura 9-30 y cuyo espectro infrarrojo muestra bandas prominentes en\(2900 \: \text{cm}^{-1}\),\(1750 \: \text{cm}^{-1}\),\(1000 \: \text{cm}^{-1}\), y\(1100 \: \text{cm}^{-1}\).
El espectro infrarrojo indica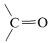 \(\left( 1750 \: \text{cm}^{-1} \right)\),\(\ce{C-H} \: \left( 2900 \: \text{cm}^{-1} \right)\), y\(\ce{C-O} \: \left( 1000 \: \text{cm}^{-1}, \: 1100 \: \text{cm}^{-1} \right)\). La posición de la banda carbonilo sugiere que probablemente sea un éster,
\(\left( 1750 \: \text{cm}^{-1} \right)\),\(\ce{C-H} \: \left( 2900 \: \text{cm}^{-1} \right)\), y\(\ce{C-O} \: \left( 1000 \: \text{cm}^{-1}, \: 1100 \: \text{cm}^{-1} \right)\). La posición de la banda carbonilo sugiere que probablemente sea un éster, . Se descarta un ácido carboxílico porque no hay signos de\(\ce{O-H}\) estiramiento.
. Se descarta un ácido carboxílico porque no hay signos de\(\ce{O-H}\) estiramiento.
El espectro de RMN muestra tres tipos de señales correspondientes a tres tipos de protones. El integral muestra que estos están en la proporción de 2:3:3. De esto, podemos concluir que son dos tipos diferentes de\(\ce{CH_3-}\) grupos y un\(\ce{-CH_2-}\) grupo.
Los desplazamientos químicos de los presuntos\(\ce{CH_3}\) grupos están en\(3.70 \: \text{ppm}\) y\(3.35 \: \text{ppm}\). Debido a que el compuesto contiene solo\(\ce{C}\)\(\ce{H}\),\(\ce{O}\), y, los datos del Cuadro 9-4 sugieren que estas resonancias surgen de\(\ce{OCH_3}\) grupos. Es probable que la resonancia de campo bajo sea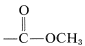 (sabemos por el infrarrojo que probablemente hay una función éster), mientras que la resonancia de campo superior es posiblemente una función éter,\(\ce{-OCH_3}\). Si juntas toda esta información, encuentras que esa\(\ce{CH_3OCH_2CO_2CH_3}\) es la única estructura posible. Para verificar si la\(\ce{CH_2}\) resonancia at\(3.9 \: \text{ppm}\) es consistente con la estructura asignada podemos calcular un valor de desplazamiento a partir de la Ecuación 9-4:
(sabemos por el infrarrojo que probablemente hay una función éster), mientras que la resonancia de campo superior es posiblemente una función éter,\(\ce{-OCH_3}\). Si juntas toda esta información, encuentras que esa\(\ce{CH_3OCH_2CO_2CH_3}\) es la única estructura posible. Para verificar si la\(\ce{CH_2}\) resonancia at\(3.9 \: \text{ppm}\) es consistente con la estructura asignada podemos calcular un valor de desplazamiento a partir de la Ecuación 9-4:
\[\begin{align} &\delta = 0.23 + \sigma_{OCH_3} + \sigma_{O=COCH_3} \\ &\delta = 0.23 + 2.36 + 1.55 = 4.14 \: \text{ppm} \end{align}\]
La concordancia entre los cambios calculados y observados no es perfecta, sino que se encuentra dentro del rango habitual de variación para la Ecuación 9-4. Podemos estar satisfechos de que la estructura asignada es correcta.
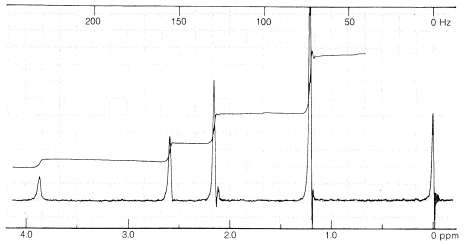
División Spin-Spin - Lo que observamos
Si nos fijamos en el espectro de RMN del etanol\(\ce{CH_3CH_2OH}\),, en la Figura 9-23, verá que la\(\ce{CH_2}\) resonancia es en realidad un grupo de cuatro líneas y la\(\ce{CH_3}\) resonancia es un grupo de tres líneas. Este patrón de tres y cuatro líneas para el agrupamiento\(\ce{CH_3CH_2X} \: \left( \ce{X} \neq \ce{H} \right)\) también es evidente en el\(220 \: \text{MHz}\) espectro de 2-metil-2-butanol (Figura 9-27) y en el\(60 \: \text{MHz}\) espectro de yoduro de etilo (Figura 9-32).
¿Por qué ciertas resonancias de protones aparecen como grupos de líneas equidistantes en lugar de resonancias simples? Los hechos son que los protones no equivalentes en los carbonos contiguos , como los derivados etílicos\(\ce{CH_3CH_2X}\), interactúan magnéticamente para “dividir” las resonancias de los demás. Esta multiplicidad de líneas producidas por la interacción mutua de núcleos magnéticos se denomina "división espín-espín “, y si bien complica los espectros de RMN, también proporciona valiosa información estructural, como veremos.
, como los derivados etílicos\(\ce{CH_3CH_2X}\), interactúan magnéticamente para “dividir” las resonancias de los demás. Esta multiplicidad de líneas producidas por la interacción mutua de núcleos magnéticos se denomina "división espín-espín “, y si bien complica los espectros de RMN, también proporciona valiosa información estructural, como veremos.
Un ejemplo de un espectro de protones complejo es el del yoduro de etilo (Figura 9-32). A una primera aproximación, los dos grupos principales de líneas aparecen como conjuntos igualmente espaciados de tres y cuatro líneas, surgidos de lo que se llama “interacciones spin-spin de primer orden”. Las cosas se complican aún más por la división adicional del patrón “tres-cuatro” del yoduro de etilo, como también se puede ver en la Figura 9-32. Esta división adicional se llama división de “segundo orden”.
Cuando hay tantas líneas presentes, ¿cómo sabemos a qué nos enfrentamos? ¿De dónde medimos el cambio químico en un complejo grupo de líneas?
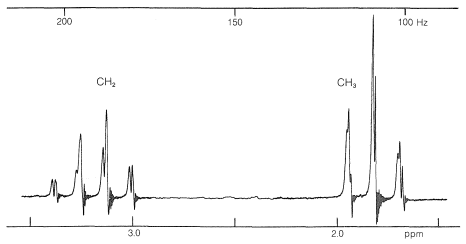
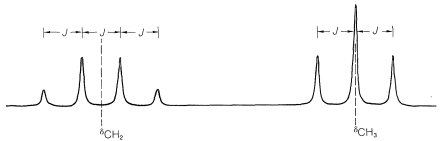
Primero, el desplazamiento químico normalmente se encuentra en el centro del grupo de líneas correspondientes a la división de primer orden. En el yoduro de etilo, el desplazamiento químico de los protones de metilo se encuentra en el centro del cuarteto:

Segundo, el desplazamiento químico puede reconocerse por el hecho de que es directamente proporcional a la frecuencia del transmisor,\(\nu\). Si doblamos\(\nu\), los desplazamientos químicos se doblan. En contraste, los splittings spin-spin de primer orden siguen siendo los mismos. Con esto queremos decir que la magnitud (in\(\text{Hz}\)) del espaciamiento entre las líneas de una resonancia dividida es independiente de la frecuencia del transmisor,\(\nu\). Este espaciado corresponde a lo que se denomina la constante de acoplamiento espín-espín, o simplemente la constante de acoplamiento, y está simbolizada por\(J\).
En tercer lugar, la división de segundo orden tiende a desaparecer al aumentar la frecuencia del transmisor. Para el yoduro de etilo (Figura 9-32), la división de segundo orden en\(60 \: \text{MHz}\) es apenas discernible en\(100 \: \text{MHz}\) y desaparece en\(200 \: \text{MHz}\). Esto también se puede ver que ocurre para el patrón de división de tres y cuatro de 2-metil-2-butanol en función de\(\nu\) (Figura 9-27).
La siguiente pregunta es ¿cómo podemos entender y predecir qué patrones de división spin-spin se observarán? ¿Y cómo nos dan información estructural? El punto importante es que a menudo se ve que la multiplicidad de líneas para protones de un desplazamiento químico dado es\(\left( n + 1 \right)\), en la que\(n\) se encuentra el número de protones en los carbonos contiguos. Por ejemplo, la\(\ce{CH_2}\) resonancia del grupo etilo del yoduro de etilo es un cuarteto de líneas debido a la interacción espín-espín con los tres protones vecinos\(\left( n = 3 \right)\) del grupo metilo. Asimismo, el\(\ce{CH_3}\) grupo es un triplete de líneas debido a las interacciones espín-espín con los dos protones\(\left( n = 2 \right)\) del grupo metileno.
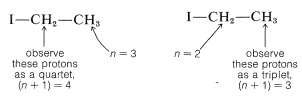
Las relaciones de las intensidades de línea en los patrones de división de espín-espín de la Figura 9-33 suelen seguir reglas simples. Un doblete aparece como dos líneas de igual intensidad; un triplete como tres líneas en la relación 1:2:1; un cuarteto como cuatro líneas en la relación 1:3:3:1; un quinteto como 1:4:6:4:1, y así sucesivamente. Las intensidades siguen los coeficientes binomiales para\(\left( x + y \right)^n\), donde\(n\) está el número de protones en el grupo de división. Así cuando\(n = 4\), tenemos\(x^4 + 4 x^3y + 6 x^2 y^2 + 4 x y^3 + y^4\), o 1:4:6:4:1.
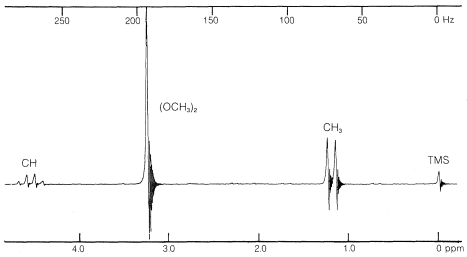
El espectro de\(\ce{(CH_3O)_2CHCH_3}\) (Figura 9-34) proporciona un excelente ejemplo de cómo la RMN muestra la presencia de protones contiguos. El doblete simétrico y el cuarteto 1:3:3:1 son típicos de la interacción entre un solo protón y un grupo adyacente de tres, es decir, . Los protones metílicos de los\(\ce{(CH_3O)}\) grupos están demasiado lejos de los demás para dar una división demostrable de espín-espín; así aparecen como una sola resonancia de seis protones.
. Los protones metílicos de los\(\ce{(CH_3O)}\) grupos están demasiado lejos de los demás para dar una división demostrable de espín-espín; así aparecen como una sola resonancia de seis protones.
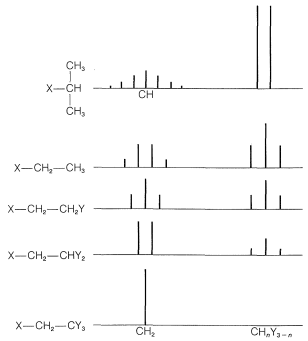
En general, la magnitud del efecto de división espín-espín de un protón sobre otro protón (o grupo de protones equivalentes) depende del número y tipo de enlaces químicos intermedios y de las relaciones espaciales entre los grupos. Para sistemas simples sin dobles enlaces y con ángulos de enlace normales, generalmente encontramos para protones no equivalentes (es decir, que tienen diferentes desplazamientos químicos):
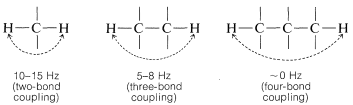
Cuando están involucrados grupos de rotación restringida o enlaces dobles y triples, se observan divisiones ampliamente divergentes. Para los dobles enlaces, los acoplamientos de dos enlaces entre dos hidrógenos no equivalentes ubicados en un extremo son característicamente pequeños, mientras que los acoplamientos de tres enlaces\(\ce{-HC=CH}-\) son más grandes, especialmente para la configuración trans:
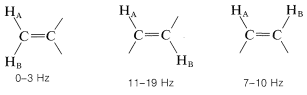
El acoplamiento a través de cuatro o más enlaces es significativo para compuestos con dobles o triples enlaces. Ejemplos de estos llamados acoplamientos de largo alcance y algunos otros valores de división útiles siguen:
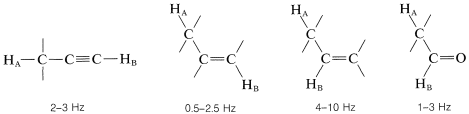
Finalmente, los protones químicamente equivalentes no dividen las resonancias entre sí.
División de protones y protones y análisis conformacional
Una característica muy importante de los acoplamientos protón-protón de tres enlaces\(\ce{H-C-C-H}\), es la forma en que dependen de la conformación en el\(\ce{C-C}\) enlace. Los valores típicos para varias conformaciones particulares son
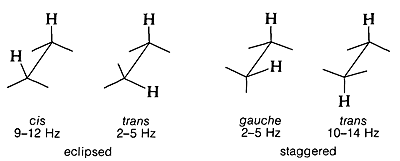
Separaciones de protones y protones e intercambio químico
Quizás te hayas preguntado por qué el protón hidroxilo del etanol produce una sola resonancia en el espectro de la Figura 9-23. Es bastante razonable esperar que el protón hidroxilo sea dividido por los protones metileno vecinos debido a que solo están separados por tres enlaces,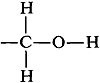 sin embargo, este acoplamiento no se observará si los protones hidroxilo se están intercambiando rápidamente entre las moléculas de etanol (Sección 9-10E). Cuando el intercambio de protones es rápido, las interacciones de espín entre los\(\ce{-OH}\) protones\(\ce{-CH_2}-\) y promedian a cero. A tipos de cambio intermedios, el acoplamiento se manifiesta a través del ensanchamiento de líneas o dando realmente múltiples líneas. Si observa los diversos espectros de etanol en la Figura 9-29, notará cómo la forma de la\(\ce{OH}\) resonancia varía de un singlete ancho a un triplete distinto.
sin embargo, este acoplamiento no se observará si los protones hidroxilo se están intercambiando rápidamente entre las moléculas de etanol (Sección 9-10E). Cuando el intercambio de protones es rápido, las interacciones de espín entre los\(\ce{-OH}\) protones\(\ce{-CH_2}-\) y promedian a cero. A tipos de cambio intermedios, el acoplamiento se manifiesta a través del ensanchamiento de líneas o dando realmente múltiples líneas. Si observa los diversos espectros de etanol en la Figura 9-29, notará cómo la forma de la\(\ce{OH}\) resonancia varía de un singlete ancho a un triplete distinto.
El intercambio químico rápido de núcleos magnéticos no es la única manera en que las interacciones espín-acoplamiento pueden promediarse a cero. El mismo efecto se puede lograr mediante una técnica conocida como doble resonancia. Para entender cómo se hace esto, considere dos protones acoplados\(\ce{H}_\text{A}\) y\(\ce{H}_\text{B}\) teniendo diferentes desplazamientos químicos. Supongamos que\(\ce{H}_\text{A}\) se irradia selectivamente a su frecuencia de resonancia\(\nu_\text{A}\) mientras que al mismo tiempo observamos la señal de resonancia de\(\ce{H}_\text{B}\). El acoplamiento entre\(\ce{H}_\text{A}\) y\(\ce{H}_\text{B}\) desaparece, y\(\ce{H}_\text{B}\) muestra una sola resonancia. ¿Por qué es así? Por irradiación de\(\ce{H}_\text{A}\), los\(\ce{H}_\text{A}\) núcleos se cambian del estado +1/2 a -1/2 y vuelven de nuevo con suficiente rapidez para que el núcleo vecino\(\ce{H}_\text{B}\) efectivamente “vea” ni un estado ni el otro. Por lo tanto, la interacción magnética entre los estados promedia a cero. Este desacoplamiento de núcleos magnéticos mediante técnicas de doble resonancia es especialmente importante en la espectroscopia de\(\ce{^{13}C}\) RMN (Sección 9-10L) pero también se utiliza para simplificar los espectros de protones mediante la eliminación selectiva de acoplamientos particulares.
Uso de la Espectroscopia de Resonancia Magnética Nuclear en Análisis Estructural
La solución de un problema típico de análisis estructural mediante métodos de RMN utiliza al menos cuatro tipos de información obtenida directamente del espectro. Son: desplazamientos químicos\(\left( \delta \right)\), intensidades de línea (áreas de señal), patrones de división de espín-espín (multiplicidades de línea) y constantes de acoplamiento\(\left( J \right)\). Ya hemos demostrado cómo se utilizan los cambios químicos en ausencia de división de espín-espín. Ahora ilustraremos cómo se pueden analizar espectros más complejos.
La Figura 9-35 muestra el espectro de RMN protónica para un compuesto de fórmula\(\ce{C_3H_6O}\). Hay tres grupos principales de líneas en\(9.8\),\(2.4\), y\(1.0 \: \text{ppm}\). Observa la multiplicidad de estos grupos antes de seguir leyendo.
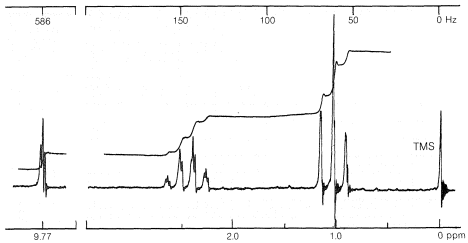
Hay varias formas de abordar un problema como este, pero probablemente la más fácil es comenzar con la integral. Las alturas relativas de la integral escalonada para los grupos principales de líneas se pueden obtener mediante un par de divisores, con regla, o con líneas horizontales como en la Figura 9-35. La integral sugiere que un hidrógeno es responsable de la resonancia en\(9.8 \: \text{ppm}\), dos hidrógenos en\(2.4 \: \text{ppm}\), y tres at\(1.0 \: \text{ppm}\). Tres hidrógenos en un solo grupo sugieren un\(\ce{CH_3}-\) grupo, y debido a que hay un patrón de división de tres a cuatro, es razonable postular\(\ce{CH_3-CH_2}-\). Restar\(\ce{C_2H_5}\) de la fórmula dada\(\ce{C_3H_6O}\) las hojas\(\ce{CHO}\), que, con valencias normales, tiene que serlo\(\ce{-CH=O}\). Por lo tanto, el espectro parece ser consistente con la estructura\(\ce{CH_3CH_2CH=O}\) (propanal) según se juzga a partir de la fórmula molecular y el patrón de división espín-espín, lo que indica el\(\ce{CH_3CH_2}-\) agrupamiento. Para estar seguros de la estructura, debemos verificarla con toda la información disponible. Primero, a partir de los turnos (Cuadro 9-4) vemos que el protón único en\(9.8 \: \text{ppm}\) encaja casi perfectamente para\(\ce{RCHO}\), la\(\ce{-CH_2C=O}\) resonancia de dos protones en\(2.4 \: \text{ppm}\) es consistente con la reportada para\(\ce{-CH_2COR}\), mientras que la línea de tres protones en\(1.0 \: \text{ppm}\) los controles con\(0.9 \: \text{ppm}\) for\(\ce{CH_3R}\).
¿Qué pasa con los acoplamientos? El patrón de tres y cuatro tiene un espaciado ligeramente superior\(7 \: \text{Hz}\), que es justo para un grupo etilo (comparar las Figuras 9-23 y 9-32). La duplicación (casi oscurecida por la división de segundo orden) de la\(\ce{-CH_2}-\) resonancia y la división de la\(\ce{-CH=O}\) resonancia en un triplete 1:2:1 indican aproximadamente un\(2\)\(\text{Hz}\) acoplamiento para el\(\ce{-CH_2-CH=O}\) grupo. Los acoplamientos de tres enlaces entre\(\ce{-CHO}\) los\(\ce{-CH_2}-\) protones adyacentes parecen ser generalmente más pequeños que\(\ce{-CH_2-CH_3}\) los acoplamientos.
Por lo general, no confiaríamos solo en la RMN en un problema de análisis estructural de este tipo, sino que buscaríamos pistas o corroboraciones de los espectros infrarrojos, electrónicos u otros espectros, así como pruebas químicas. En capítulos posteriores tendremos muchos problemas que se verán facilitados por el uso tanto de espectros de RMN como de infrarrojos. Otro ejemplo trabajado ilustrará el enfoque.
Un compuesto tiene la composición\(\ce{C_3H_3Br}\) y da los espectros de resonancia magnética infrarroja y nuclear mostrados en la Figura 9-36. El problema es cómo utilizar esta información para deducir la estructura del compuesto. La fórmula molecular nos dice el número y tipo de átomos y el número de múltiples enlaces o anillos. Las fórmulas del\(\ce{C_3}\) hidrocarburo correspondiente sin el bromo serían\(\ce{C_3H_4}\), o cuatro hidrógenos menores que el alcano saturado\(\ce{C_3H_8}\). Esto significa que debe haber dos dobles enlaces o el equivalente: un triple enlace o un anillo y un doble enlace. \(^{14}\)Porque a partir de la fórmula sospechamos de la instauración, deberíamos comprobarlo con el espectro infrarrojo. Hay una banda at\(2120 \: \text{cm}^{-1}\), que es indicativa de un\(\ce{-C \equiv C}-\) grupo asimétricamente sustituido (Cuadro 9-2). La banda fuerte y afilada a\(3300 \: \text{cm}^{-1}\) más lejos nos dice que la sustancia es un 1-alquino\(\ce{-C \equiv C-H}\).
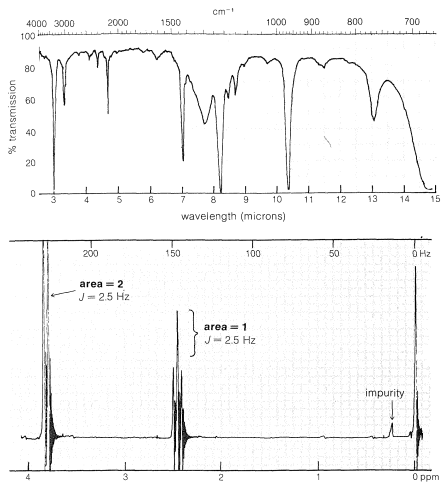
El espectro de RMN de protones muestra que solo hay dos grupos principales de líneas: un doblete de dos protones en\(3.85 \: \text{ppm}\) y un triplete de un protón en\(2.45 \: \text{ppm}\). El patrón de división de dos a tres combinado con la relación protónica 2:1 sugiere un\(\ce{CH_2}\) grupo acoplado con un\(\ce{CH}\) grupo. La estructura debe ser un 3-bromo-propiino,\(\ce{BrCH_2C \equiv CH}\). Para confirmar la asignación, se deben verificar los cambios químicos (Cuadro 9-4). El\(\ce{\equiv C-H}\) at\(2.45 \: \text{ppm}\) concuerda bien con el valor tabulado de\(2.5 \: \text{ppm}\). No hay datos tabulados para\(\ce{-C \equiv C-CH_2Br}\) pero el desplazamiento observado en\(3.85 \: \text{ppm}\) se encuentra en campos ligeramente menores que los tabulados\(3.33 \: \text{ppm}\) para\(\ce{-CH_2Br}\). Esto se espera por el triple vínculo. La correlación de la Ecuación 9-4 predice un valor de\(4.0 \: \text{ppm}\).
Muy a menudo, un protón se acoplará por giro a dos o más protones diferentes, y los acoplamientos no son necesariamente los mismos. Cuando esto sucede, el espectro resultante puede ser bastante complejo, como muestra nuestro siguiente ejemplo.
Un compuesto\(\ce{C_9H_{10}}\) da el espectro de RMN de la Figura 9-37. Claramente hay cuatro tipos de protones en la molécula en\(\delta = 7.28 \: \text{ppm}\),\(5.35 \: \text{ppm}\),\(5.11 \: \text{ppm}\), y\(1.81 \: \text{ppm}\). Aunque no se muestra la integral, los principales grupos de líneas tienen intensidades desde el campo bajo hasta el campo alto en la proporción de 5:1:1:3.
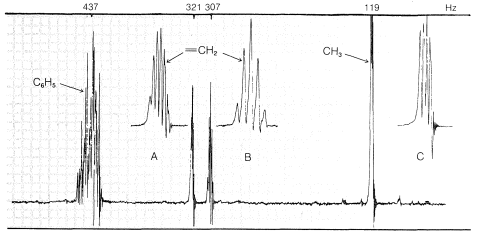
La señal de cinco protones en\(7.28 \: \text{ppm}\) es típica de un grupo fenilo,\(\ce{C_6H_5}\), y las señales de un protón en\(5.35\) y\(5.11 \: \text{ppm}\) están en la región para protones alquénicos,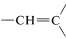 . La señal de tres protones en\(1.81 \: \text{ppm}\) es típica de un grupo metilo en un doble enlace carbono-carbono,
. La señal de tres protones en\(1.81 \: \text{ppm}\) es típica de un grupo metilo en un doble enlace carbono-carbono,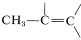 .
.
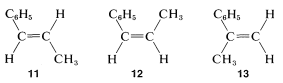
Sólo hay tres formas de armar un anillo de fenilo,, y dos\(\ce{HC=}\) protones tales que suman\(\ce{C_9H_{10}}\). Ellos son
El acoplamiento entre A y B (designado por la constante\(J_\text{AB}\)) debe dar cuatro líneas, dos para A y dos para B, como se muestra en la Figura 9-38. Debido a que A y B también están acoplados a los tres hidrógenos del grupo metilo (C), cada una de las cuatro líneas correspondientes se dividirá aún más (en cuartetos 1:3:3:1).\(J_\text{AB}\) Si\(J_\text{AC} \neq J_\text{BC}\), entonces el espaciado de las líneas en los dos conjuntos de cuartetos no será el mismo.
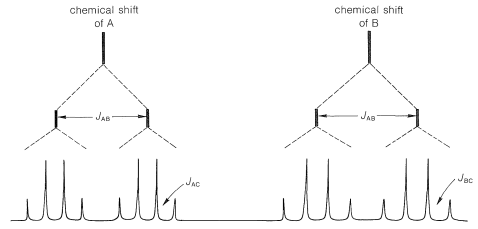
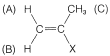 where\(J_\text{AB} \gg J_\text{B} > J_\text{AC}\).
where\(J_\text{AB} \gg J_\text{B} > J_\text{AC}\).De acuerdo con el análisis anterior, el número máximo de líneas observables para las resonancias A y B es de dieciséis (8 para A y 8 para B). De hecho, solo once son visibles (6 para A y 5 para B), lo que significa que algunas de las dieciséis líneas posibles deben solaparse. Sin examinar todas las posibilidades, podemos ver que la situación real se puede reproducir si\(J_\text{AB} \cong J_\text{BC} = 2J_\text{AC}\). La única estructura con la que es consistente\(J_\text{AB} = 1.5 \: \text{Hz}\) es\(13\), o 2-fenilpropeno; las otras posibilidades están excluidas porque\(J_\text{AB}\) deben ser sobre\(10 \: \text{Hz}\) para\(12\) y\(16 \: \text{Hz}\) para\(11\).
Efectos de cambio químico en la división Spin-Spin
La\(n + 1\) regla simple para predecir la multiplicidad de señales de protones acopladas por giro a menudo se rompe cada vez que la diferencia de desplazamiento químico entre los protones en diferentes grupos se vuelve comparable a las constantes de acoplamiento para la interacción magnética entre los grupos. En estas circunstancias, puede esperar ver más líneas, o líneas en diferentes posiciones con diferentes intensidades, de las predichas a partir del simple tratamiento de primer orden. Un ejemplo es el efecto de cambiar el desplazamiento químico en un espectro de dos protones con\(J = 10 \: \text{Hz}\) (Figura 9-44).
Vemos en la Figura 9-44 que incluso cuando el desplazamiento es 7.5 veces mayor que el acoplamiento, las líneas exteriores son más débiles que las líneas internas. Este tipo general de asimetría de intensidades de línea también es evidente en el espectro de yoduro de etilo (Figura 9-32), en el que las líneas de cada grupo se parecen más a 0. 7:2. 5:3. 5:1 .3 y 1. 2:2. 0:0 .8, en lugar de las proporciones 1:3:3:1 y 1:2:1 predichas a partir del tratamiento de primer orden. La asimetría es tal que dos grupos de líneas que están conectados por división de espín-espín en efecto “apuntan” entre sí: las líneas en el “interior” del patrón son más fuertes de lo previsto desde el tratamiento de primer orden, mientras que las del “exterior” son más débiles. El efecto se puede poner en práctica, como se ilustra en el siguiente ejercicio.
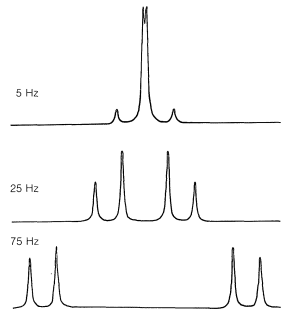
Explicar el efecto de los cambios químicos en la división de segundo orden está fuera del alcance de este libro. De hecho, realmente no hemos explicado la división de primer orden, aunque más sobre este tema se encontrará en la Sección 27-3. Pero independientemente de cuántas líneas aparezcan en un espectro de RMN complejo, se pueden racionalizar en términos de los desplazamientos químicos, las constantes de acoplamiento y los efectos de intercambio. Además, las intensidades de señal globales permanecen proporcionales al número de protones que dan lugar a las señales.
Cuando hay muchos hidrógenos y pequeñas diferencias de desplazamiento químico, como en los alcanos, los espectros de RMN de protones pueden tener tantas líneas de resonancia estrechamente espaciadas que se fusionan para dar una serie de picos suaves, más o menos característicos. El espectro protónico del octano (Figura 9-46a) es un excelente ejemplo de este tipo de espectro. A menudo se puede obtener información útil a partir de espectros tales como la relación de\(\ce{CH_3}\)\(\ce{CH_2}\)::\(\ce{CH}\) mediante la investigación de las integrales en el rango de absorciones de protones alcanos. La Figura 9-46 ilustra cómo se puede hacer esto para octano y 2,2,4-trimetilpentano.

\ (n\) carbonos. Para el octano (a), la relación integral es 1:2 o 6:12. </em">
Espectroscopia de Resonancia Magnética Nuclear Carbon-13
En los últimos años, la espectroscopia\(\ce{^{13}C}\) de\(\ce{^{13}C}\) RMN utilizando abundancia natural se\(\left( 1.1 \% \right)\) ha convertido en una herramienta importante para el análisis estructural orgánico. Que esto no sucedió antes es porque\(\ce{^{13}C}\) tiene un momento magnético mucho menor que\(\ce{^1H}\) y el momento pequeño combinado con la pequeña abundancia natural significa que\(\ce{^{13}C}\) es más difícil de detectar en la RMN que\(\ce{^1H}\) por un factor de 5700. Esta es una gran diferencia y se puede poner en el contexto adecuado de la siguiente manera. Supongamos que dos personas están hablando en una habitación ruidosa y una está tratando de escuchar a la otra. El pedido común es “hablar más fuerte”. Si esto no es posible entonces la petición es “decirlo de nuevo” o “hablar más despacio”. Cualquiera de estas últimas solicitudes equivale a una integración de señal versus ruido y lleva tiempo. La mejora en señal-ruido para una comunicación dada se logra como la raíz cuadrada del tiempo de comunicación. En base al tiempo crucial, las señales de\(\ce{^{13}C}\) RMN requieren\(\left( 5700 \right)^2 \cong 30,000,000\) veces más tiempo para obtener la misma relación señal/ruido que en la\(\ce{^1H}\) RMN para el mismo número de núcleos por unidad de volumen. Esto es un problema.
Las mejoras electrónicas y el uso de la teoría de la comunicación, con énfasis en la técnica de “decirlo de nuevo”, han proporcionado los medios para obtener\(\ce{^{13}C}\) espectros de rutina incluso para soluciones bastante diluidas de moléculas bastante complejas.
Algunos de los mismos tipos de efectos estructurales son importantes para los cambios\(\ce{^{13}C}\) químicos que para los desplazamientos químicos de protones (Sección 9-10E). Por ejemplo, existe un paralelo similar entre las diferencias de\(\ce{^{13}C}\) desplazamiento en compuestos del tipo\(\ce{CH_3-CH_2-X}\) y electronegatividad (Figura 9-47) como entre los correspondientes desplazamientos de protones y electronegatividad (Figura 9-28). Es importante notar que\(\ce{^{13}C}\) los turnos en\(\text{ppm}\) las unidades son mucho mayores que los de los protones. Esto se debe a que el carbono usa\(p\) orbitales para formar enlaces, mientras que el hidrógeno usa\(s\) orbitales. Por lo tanto, esperaremos encontrar los núcleos de otros elementos que utilizan\(p\) orbitales en la unión, como\(\ce{^{15}N}\),\(\ce{^{19}F}\), y\(\ce{^{31}P}\), también tendrán desplazamientos mayores que para los protones, como en efecto lo hacen.
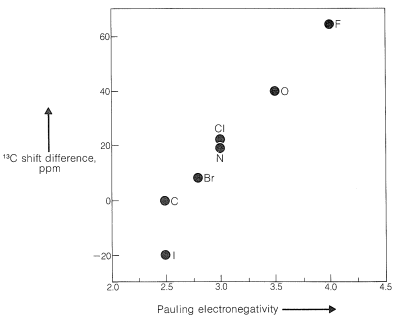
\ (\ ce {CH_3CH_2X}\) derivadas en función de la electronegatividad de Pauling. Los carbonos metílicos de los\(\ce{CH_3CH_2X}\) derivados son\(15\) -\(22 \: \text{ppm}\) campo abajo\(\ce{^{13}C}\) del TMS. </em">
En la\(\ce{^{13}C}\) Figura 9-48 se muestra una aplicación estructural de la\(\ce{^1H}\) RMN, que muestra su potencia en un área donde la RMN es indecisa. Aquí, vemos las\(\ce{^{13}C}\) resonancias de campo alto de una sustancia conocida de diversas maneras como Coumadin, o la sal sódica de warfarina\(14\), que es ampliamente utilizada como anticoagulante sanguíneo en el tratamiento de enfermedades como la flebitis. También tiene una utilidad sustancial como veneno para ratas debido a su acción anticoagulante.
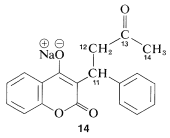
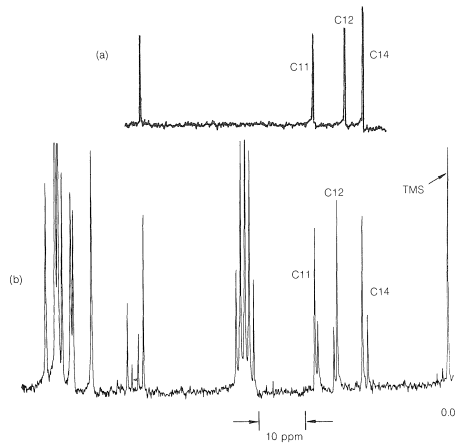
No hay indicación alguna de anormalidad en los desplazamientos químicos de los carbonos 11, 12 y 14 mostrados en la Figura 9-48a. Además, existe una resonancia de campo abajo\(216.5 \: \text{ppm}\) de los carbonos del TMS (no mostrado en la Figura 9-48a) que es típica de un\(\ce{C=O}\) carbono correspondiente a C13. Cuando\(14\) se trata con ácido, se espera que se forme el producto (warfarina) de estructura, el cual debe tener un\(\ce{^{13}C}\) espectro muy parecido\(15\) al que se muestra en la Figura 9-48a.

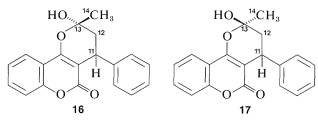
Los\(\ce{^{13}C}\) datos indican claramente que la warfarina no está\(15\) en solución sino que es una mezcla de dos diastereómeros (\(16\)y\(17\), llamados hemicetales cíclicos) resultante de la adición del\(\ce{-OH}\) grupo de\(15\) al\(\ce{C=O}\) enlace:
Este es un ejemplo del poder de la\(\ce{^{13}C}\) RMN para resolver problemas estructurales sutiles.
\(^8\)Aunque los isótopos principales de\(Cl\)\(Br\), y\(I\) tienen propiedades magnéticas, debido al carácter especial de todos estos isótopos, actúan en compuestos orgánicos como si fueran no magnéticos.
\(^9\)La resonancia en el sentido aquí utilizado significa que la absorción de radiofrecuencia tiene lugar a frecuencias de “resonancia” especificadas. Sin embargo, verás que casi todas las formas de espectroscopia que discutimos en este libro implican absorción de “resonancia” en el mismo sentido.
\(^{10}\)Aquí,\(\gamma\) está en\(\text{Hz}\) per gauss; los físicos suelen definir\(\gamma\) en radianes por segundo por gauss.
\(^{11}\)Del prefijo griego dia significado a través, a través. Lo opuesto de diamagnético es paramagnético; para significando al lado. Utilizaremos este término más adelante.
\(^{12}\)Además de dar una mejor separación de las líneas y espectros más claros, ir a campos superiores también tiene el efecto beneficioso de aumentar las proporciones de los núcleos en el\(+ \frac{1}{2}\) estado, dando así resonancias más intensas, más fáciles de detectar.
\(^{13}\)Muchos otros valores de desplazamiento de protones están disponibles en NMR Spectra Catalog, Volumen 1 y 2, Varian Associates, Palo Alto, Calif., 1962, 1963.
\(^{14}\)Si estuvieran presentes dos anillos, esto también daría cuatro hidrógenos menos que el alcano. Sin embargo, no son posibles dos anillos con solo tres carbonos.
Referencias
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."