
18.3: Algunas propiedades químicas de los ácidos carboxílicos
- Page ID
- 72924

La mayoría de las reacciones de los ácidos carboxílicos pertenecen a una de las cuatro clases principales, dependiendo del punto en la molécula donde se produzca la reacción.
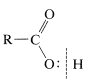
a. Reacciones que involucran el\(\ce{O-H}\) enlace: estas incluyen la disociación ácida y las reacciones solvolíticas.
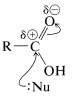
b. Reacciones en el enlace carbonilo, la mayoría de las cuales implican el ataque de un nucleófilo\(: \ce{Nu}\) sobre el carbono carbonilo con posterior escisión de un\(\ce{C-O}\) enlace. Los ejemplos son esterificación, formación de cloruro de acilo y reducción con hidruros.
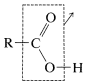
c. Descarboxilación - son reacciones en las que el\(\ce{R-C}\) enlace se rompe de tal manera que\(\ce{CO_2}\) se pierde y\(\ce{R-H}\) se forma.
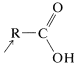
d. Sustitución en el\(\ce{R}\) grupo - las sustituciones por hidrógeno o halógeno en el carbono 2 son especialmente importantes.
Destacaremos la manera en que la química de los ácidos carboxílicos en cada una de estas categorías puede correlacionarse con los principios esbozados en capítulos anteriores.
Disociación de Ácidos Carboxílicos. El efecto de resonancia
En comparación con los ácidos minerales como los ácidos clorhídrico, perclórico, nítrico y sulfúrico, los ácidos carboxílicos,\(\ce{CH_3(CH_2)}_n \ce{CO_2H}\), son débiles. El grado de disociación en solución acuosa es relativamente pequeño, siendo las constantes de acidez\(K_\text{a}\), aproximadamente\(10^{-5}\) (ver Cuadro 18-1).
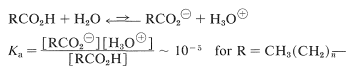
A pesar de que los ácidos carboxílicos son ácidos débiles, son muchos órdenes de magnitud más fuertes que los alcoholes correspondientes,\(\ce{CH_3(CH_2)}_n \ce{CH_2OH}\). Así el\(K_\text{a}\) del ácido etanoico\(\ce{CH_2CO_2H}\),, es\(10^{11}\) veces mayor que el del etanol,\(\ce{CH_3CH_2OH}\).
La acidez del grupo carboxilo surge, al menos en parte, de la naturaleza polar del grupo carbonilo, cuya polaridad puede atribuirse a contribuciones de la estructura
Para un grupo carboxilo, estas estructuras y una posibilidad adicional se muestran mediante\(1a\)\(1b\), y\(1c\):
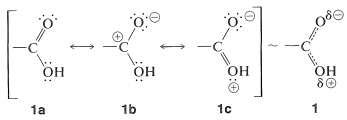
Si bien la estructura no cargada\(1a\),, es de gran importancia, estructura\(1b\) y\(1c\) hace contribuciones significativas. La estabilización es sustancial y los ácidos carboxílicos son más estables de lo que se esperaría, al sumar completamente sus energías de enlace\(18 \: \text{kcal mol}^{-1}\).
La energía de estabilización del anión carboxilato es sustancialmente mayor que la del ácido, debido a que el anión es un híbrido de resonancia de dos estructuras energéticamente equivalentes,\(2a\) y\(2b\), mientras que el ácido está representado por un híbrido de estructuras no equivalentes, \(1a\)a través de\(1c\):
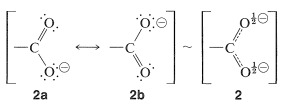
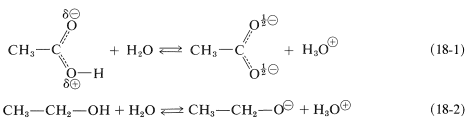
Las reglas para el estrés de resonancia que se espera la mayor estabilización cuando las estructuras contribuyentes son equivalentes (Sección 6-5B). Por lo tanto, podemos concluir que la energía de resonancia de un anión carboxilato debe ser mayor que la del ácido correspondiente. En consecuencia podemos decir que existe una “fuerza impulsora” (una ganancia en la estabilidad) que promueve la disociación de los ácidos carboxílicos. El hecho de que los alcoholes sean ácidos mucho más débiles que los ácidos carboxílicos puede atribuirse a la falta de estabilización de los iones alcóxido en comparación con la de los aniones carboxilato. La diferencia de energía correspondiente a la disociación de un ácido carboxílico (Ecuación 18-1) con respecto a la de un alcohol (Ecuación 18-2) en realidad asciende a aproximadamente\(15 \: \text{kcal mol}^{-1}\):
El efecto inductivo y las fuerzas ácidas
Es posible que hayas notado que existen diferencias considerables entre las fuerzas de algunos de los ácidos enumerados en el Cuadro 18-1. El ácido metanoico y casi todos los ácidos etanoicos sustituidos son más fuertes que el ácido etanoico. De hecho, el ácido trifluoroetanoico es similar en fuerza al ácido clorhídrico. Los grupos sustituyentes claramente pueden tener un profundo efecto sobre la fuerza ácida por lo que comúnmente se llama el efecto inductivo, un efecto relacionado con la electronegatividad del sustituyente. El efecto inductivo es diferente de los efectos de resonancia discutidos en la Sección 18-2A en que se asocia con la sustitución en los átomos de carbono saturados de la cadena. El efecto inductivo del sustituyente hace que el ácido sea más fuerte o más débil (en relación con el ácido no substituido), dependiendo de si el sustituyente es atrayente de electrones o donador de electrones en relación con el hidrógeno.
La escala de electronegatividad (Sección 10-4B) muestra que el cloro atrae más electrones que el hidrógeno, y el ácido cloroetanoico es un ácido 80 veces más fuerte que el propio ácido etanoico. La sustitución por más cloros potencia la acidez. El ácido dicloroetanoico es 3000 veces y el ácido tricloroetanoico es 5000 veces más ácido que el ácido etanoico. Alejar la posición de sustitución a lo largo de la cadena del grupo carboxilo hace que el efecto sea más pequeño, y el ácido 4-clorobutanoico es solo un ácido dos veces más fuerte que el ácido butanoico (Cuadro 18-1).
Se puede considerar que el efecto inductivo del sustituyente se transmite al grupo carboxilo de dos maneras bastante diferentes. Con mayor frecuencia, se considera que el sustituyente causa cambios en las distribuciones promedio de los electrones de enlace a lo largo de la cadena de átomos entre este y el protón carboxilo. Esto produce una sucesión de desplazamientos de electrones a lo largo de la cadena, lo que, para un sustituyente que atrae electrones, aumenta la fuerza ácida al hacer que sea más energéticamente factible que el\(\ce{-OH}\) hidrógeno del grupo carboxilo salga como protón:
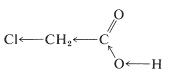
Muchos otros grupos además de los halógenos exhiben efectos inductivos potenciadores de ácido atrayentes de electrones. Entre estos se encuentran nitro\(\left( \ce{-NO_2} \right)\), metoxi\(\left( \ce{CH_3O} \right)\), carbonilo (\(\ce{RCOR'}\), como en aldehídos, cetonas, ácidos, ésteres y amidas), ciano o nitrilo\(\left( \ce{-C \equiv N} \right)\), y trialquilamonio\(\left( \ce{R_3} \overset{\oplus}{\ce{N}} \ce{-} \right)\). Los grupos alquilo -metilo, etilo, isopropilo, etc.- son los únicos sustitutivos enumerados en la Tabla 18-1 que debilitan el ácido en relación con el hidrógeno (como puede verse comparando los\(K_\text{a}\) valores de los ácidos de cadena más larga con los de los ácidos metanoico y etanoico). Podemos tomar esto en el sentido de que los grupos alquilo liberan electrones al grupo carboxilo.
La interpretación electrostática de las resistencias ácidas
El otro modo posible de transmisión del efecto polar de un grupo sustituyente es uno puramente electrostático, a veces llamado el “efecto de campo”, en el que el dipolo del sustituyente produce un campo electrostático en el protón carboxilo, lo que ayuda o dificulta la ionización dependiendo de la forma en que el dipolo está orientado con respecto al grupo carboxilo. Es más fácil visualizar cómo opera la teoría electrostática considerando un protón a medio camino entre dos aniones carboxilato bien separados y decidiendo con cuál puede combinarse el protón de manera más favorable. El más favorable corresponderá al anión carboxilato más básico y al ácido carboxílico más débil. Con\(\ce{CH_3CO_2^-}\) y\(\ce{ClCH_2CO_2^-}\) como ejemplos, y recordando que el\(\ce{Cl-C}\) vínculo está polarizado como\(\overset{\delta \ominus}{\ce{Cl}} \ce{-} \overset{\delta \oplus}{\ce{C}}\), podemos escribir:
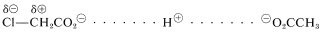
El protón será atraído por ambos\(\ce{-CO_2^-}\) grupos y las diferencias en la energía electrostática de su combinación con uno u otro de los grupos carboxilato dependerán de la influencia del\(\overset{\delta \ominus}{\ce{Cl}} \ce{-} \overset{\delta \oplus}{\ce{C}} \ce{H_2}\) dipolo. El\(\overset{\delta \ominus}{\ce{Cl}}\) final del dipolo atraerá al protón, pero el\(\overset{\delta \oplus}{\ce{C}} \ce{H_2}\) final lo repelerá. El efecto de repulsión será más importante porque el\(\overset{\delta \oplus}{\ce{C}} \ce{H_2}\) está más cerca\(\overset{\delta \ominus}{\ce{Cl}}\) que del punto final de unión del protón al oxígeno carboxilato. \(^2\)Así, el protón irá más favorablemente a\(\ce{CH_3CO_2^-}\) que a\(\ce{ClCH_2CO_2^-}\), lo que significa que\(\ce{ClCH_2CO_2H}\) es un ácido más fuerte que\(\ce{CH_3CO_2H}\).
La aplicación cuantitativa de la teoría electrostática al efecto de los sustituyentes sobre la ionización de ácidos carboxílicos, como el ácido cloroetanoico, se vuelve difícil por el hecho de que el\(\overset{\delta \ominus}{\ce{Cl}} \ce{-} \overset{\delta \oplus}{\ce{C}} \ce{H_2}\) enlace polarizado y el protón no pueden tratarse como si fueran cargas puntuales en vacío. Se debe tomar en cuenta el asunto interviniente y circundante. Esto es especialmente cierto para las soluciones de agua porque una molécula de un ácido orgánico en el agua es una cavidad de baja constante dieléctrica sumergida en un medio de alta constante dieléctrica. Por lo tanto, cuando ocurre la ionización el protón va desde el interior de la cavidad a través del límite hacia el agua. Al mismo tiempo, la naturaleza de la cavidad debe cambiar porque entonces contiene un anión, no una molécula neutra.
¿Qué papel juega la entropía en la disociación de los ácidos carboxílicos?
Se ha discutido la influencia de los sustituyentes en las fuerzas ácidas de los ácidos carboxílicos simples como si el efecto electrostático completo del sustituyente se ejerciera únicamente sobre\(\Delta H\) la ionización. Sin embargo, el cuidadoso análisis termodinámico de las acidedades en solución acuosa muestra que los efectos de entropía (Sección 4-4B) son muy importantes. Esto puede parecer sorprendente porque los efectos de entropía deben ser pequeños para las fuerzas ácidas relativas, lo que puede ser evaluado por las constantes para equilibrios simples como la Ecuación 18-3, en la que (1) hay el mismo número de moléculas en cada lado de la ecuación, y (2) las restricciones sobre la especie involucrados apenas parecen diferentes de un lado de la ecuación al otro:
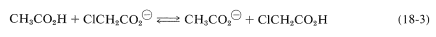
Los efectos de entropía asociados a estos equilibrios tienen que ver con el participante “invisible”, el agua, que se involucra de manera íntima, aunque por convención la omitimos de ecuaciones como 18-3. La solvatación de iones impone restricciones a las moléculas de agua, y los mismos efectos electrostáticos que cambian la facilidad de eliminación del protón actúan para cambiar el grado y la naturaleza de la solvatación, requiriendo así la consideración de los efectos de entropía en los equilibrios.
Si los efectos de la entropía de solvatación son importantes, ¿cómo podemos justificar el uso de la teoría electrostática simple para explicar los cambios en las fuerzas ácidas producidas por los sustituyentes La respuesta radica en\(\Delta G\); cualesquiera que sean los efectos electrostáticos que estén haciendo para equilibrar entre\(\Delta H\) y\(\Delta S\), es\(\Delta G\) que determina la constante de equilibrio y sigue de manera\(\Delta G\) bastante consistente las predicciones de consideraciones electrostáticas simples. Además, las fuerzas ácidas relativas de una serie de ácidos etanoicos sustituidos se han determinado en la fase gaseosa por resonancia ión-ciclotrón (Sección 27-8), bajo condiciones en las que la asociación y los efectos del disolvente están ausentes (Sección 11-8A). En la fase gaseosa, los efectos de entropía son pequeños y las acideces relativas están en el orden esperado de la escala de electronegatividad, proporcionada en correctores para el efecto de tamaño iónico que encontramos previamente con respecto a las acidedades en fase gaseosa de alquinos y alcoholes (Sección 11-8B y 15-4A). Así, el ácido fluoroetanoico es más débil que el ácido cloroetanoico en la fase gaseosa, mientras que lo contrario es cierto en solución acuosa. La diferencia puede deberse simplemente al hecho de que los iones más grandes son en general más estables que los iones más pequeños en la fase gaseosa.
Ácidos Carboxílicos como Bases
Además de sus propiedades ácidas, los ácidos carboxílicos también pueden actuar como bases débiles cuando el oxígeno carbonílico acepta un protón de un ácido fuerte, como\(\ce{H_2SO_4}\)\(\ce{HClO_4}\), o\(\ce{HSbF_6}\) en\(\ce{SO_2}\) (Ecuación 18-4). Dicha protonación es un paso importante en la esterificación catalizada por ácido, como se discute en la Sección 15-4D:
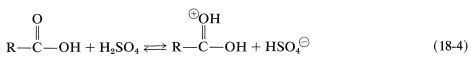
Un protón también puede agregar oxígeno al hidroxilo (Ecuación 18-5). El ácido conjugado resultante normalmente es menos favorable que su isómero con el protón en el grupo carbonilo. Sin embargo, este ácido conjugado juega un papel en la esterificación cuando el\(\ce{R}\) grupo es particularmente voluminoso y, además, tiene propiedades donadoras de electrones, favoreciendo así la ionización a un carbocatión acilo (como en la Ecuación 18-6; ver también Sección 18-3A):

Sales de Ácidos Carboxílicos como Jabones. Formación Micelar
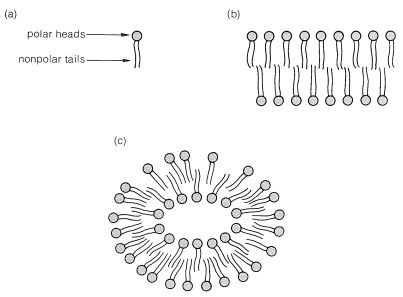
Los ácidos carboxílicos tienen un importante uso práctico en forma de sus sales metálicas como jabones. Hemos mencionado cómo las grasas, que son ésteres de 1,2,3-propanotriol (glicerilo) de ácidos de cadena larga, pueden hidrolizarse con álcalis para dar las sales carboxilato correspondientes. Se ha sabido ya en la época romana (Plinio) que tales sustancias tienen valor para fines de limpieza. \(^3\)Estas sales tienen una interacción complicada con el agua porque son muy polares en el extremo salino de la molécula y muy no polares en el extremo hidrocarbonado de cadena larga de la molécula. Estos extremos hidrocarbonados no son compatibles con un disolvente polar como el agua. \(^4\)
Cuando se ponen cantidades diminutas de jabones en el agua, en lugar de formar soluciones simples, las moléculas se concentran en la superficie del agua, con los extremos salinos pegándose al agua y las cadenas de hidrocarburos formando una capa en la superficie. Esta disposición reduce en gran medida la tensión superficial del agua y contribuye a las sorprendentes propiedades de las películas y burbujas de jabón. A concentraciones más altas, las soluciones se vuelven turbias como resultado de la formación de micelas. Las micelas son agregados considerables de moléculas de jabón, en donde las cadenas de hidrocarburos forman una región de baja polaridad que se estabiliza al tener los extremos de sal polar de las moléculas en contacto con el agua (Figura 18-4).
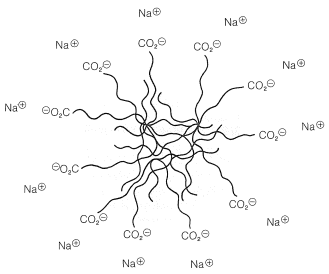
La acción limpiadora del jabón se debe en parte a la forma en que el jabón disminuye la tensión superficial del agua, ayudándola así a penetrar en los tejidos, y también a la capacidad de las micelas para solubilizar aceites y grasas al llevarlos a sus regiones hidrocarbonadas.
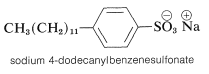
Una desventaja importante de los jabones de carboxilato simples es que se combinan con los iones calcio y magnesio normalmente presentes en la mayoría del agua del grifo para formar escoria insolubles, que interfieren con el proceso de limpieza. Se han desarrollado muchos de los llamados detergentes que no tienen esta desventaja; un ejemplo es el 4-dodecanilbencenosulfonato de sodio, cuyas sales de calcio y magnesio son solubles en agua.
Cuando las sales de carboxilato se ponen en disolventes no polares, a menudo se forman micelas inversas, donde las partes polares de las moléculas están en el interior y las partes no polares están en el exterior.
Se han observado diferencias pronunciadas para las tasas de reacciones químicas en micelas en comparación con el agua pura. Por ejemplo, la solvolisis del sulfonato de 1-metilheptilo\(5\), en solución acuosa diluida avanza 70 veces más lenta cuando\(\left( \overset{\oplus}{\ce{Na}} \overset{\ominus}{\ce{O}} \ce{SO_3C_{12}H_{25}} \right)\) se agrega suficiente dodecanilsulfato de sodio para proporcionar aproximadamente el doble de iones de sulfato de dodecanilo en el estado micelar que hay moléculas de\(5\) presente:
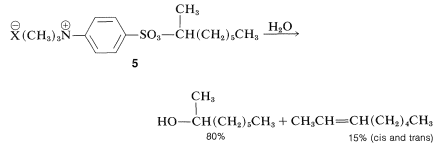
Esta ralentización de la reacción de solvolisis por parte del alquilsulfato requiere que\(5\) sea encarcelado casi por completo por las micelas, debido a que esa parte de\(5\) libre en agua se hidrolizaría rápidamente. Un resultado importante está en la estereoquímica de la reacción, que cambia de\(100\%\) inversión con ópticamente activo\(5\) en agua pura a solo\(56\%\) inversión en las micelas. Micelas de polaridad opuesta, hechas de bromuro de hexadeciltrimetilamonio\(\ce{C_{16}H_{33}} \overset{\oplus}{\ce{N}} \ce{(CH_3)_3} \overset{\ominus}{\ce{Br}}\), no tienen efecto sobre la tasa de solvolisis de\(5\).
Se han realizado estudios de este tipo en varios sistemas y son de gran interés por la luz que pueden arrojar sobre la estructura y función de las membranas biológicas.
Existe un parecido cercano entre las sales de ácidos grasos y los fosfolípidos en que ambos poseen largas colas de hidrocarburos y una cabeza polar. Los fosfolípidos también se agregan en un medio polar para formar micelas y estructuras bicapa continuas como se muestra en la Figura 18-5. La estructura lipídica bicapa es muy importante para la función autosellante de las membranas y su impermeabilidad a moléculas muy polares.
\(^2\)La energía electrostática involucrada en el salmuera de dos cargas de una distancia\(r_1\) a una\(r_2\) distancia entre sí viene dada por\(\left( e_1 e_2/D \right) \left( 1/r_1 - 1/r_2 \right)\), donde\(e_1\) y\(e_2\) son la magnitud de las cargas y\(D\) es la constante dieléctrica del medio (Sección 8-7F). Si\(e_1\) y\(e_2\) tienen el mismo signo, la energía es positiva, y con signos opuestos la energía es negativa. Para la calibración, la energía electrostática resultante de llevar una carga positiva desde una gran distancia\(\left( 1/r_1 \sim 0 \right)\), hasta una carga negativa a una distancia de\(1\) Å en vacío\(\left( D = 1 \right)\) es\(-335 \: \text{kcal mol}^{-1}\).
\(^3\)Hasta el siglo XIX los jabones se elaboraban hirviendo grasas animales o vegetales con cenizas de madera, las cuales contienen, además de sílice, cantidades considerables de potasio carbonatado. La mezcla resultante de sales de carboxilato de potasio da un jabón “blando”, y este puede convertirse en un jabón “duro” por tratamiento con exceso\(\ce{NaCl}\), que forma las sales de carboxilato de sodio menos solubles. El\(\ce{KCl}\) formado entra en la fase acuosa.
\(^4\)Uno bien podría preguntarse por qué las moléculas de jabón no simplemente cristalizan de la solución acuosa si las cadenas de hidrocarburos son incompatibles con el agua. Sin embargo, no es probable que el empaquetamiento cristalino de las partes polares de sal de la molécula sea muy compatible con las partes hidrocarbonadas y, además, la mayoría de los jabones son sales de mezclas de ácidos alifáticos y esto apenas ayuda a que se produzca la cristalización.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."