
21.3: Comparación de los métodos de Resonancia y Molecular-Orbital
- Page ID
- 72809

El enlace de par de electrones
En esta sección, esbozaremos las similitudes y diferencias en los enfoques de resonancia (o enlace de valencia, VB) y molecular-orbital (MO) para los enlaces de pares de electrones. Ambos métodos normalmente comienzan con orbitales atómicos\(1s\),\(2s\)\(2p\), y así sucesivamente, de los tipos discutidos en la Sección 6-1. Donde difieren los métodos es en cómo se utilizan estos orbitales. Para un enlace entre dos átomos, el procedimiento MO combina (o mezcla) dos orbitales atómicos, uno de cada átomo, con cuenta adecuada de la fase orbital (Sección 6-2) para obtener dos orbitales moleculares, uno de baja energía y otro de mayor energía. Los orbitales atómicos pueden ser orbitales puros o híbridos (Secciones 6-1 y 6-4). En la Figura 21-2, se muestran los resultados de combinar los\(1s\) orbitales de hidrógeno. El cálculo para el estado más estable procede determinando la energía del sistema cuando dos electrones emparejados están en el orbital molecular de baja energía. La energía de unión es la diferencia entre la energía así calculada y las energías de los átomos separados. Debido a que el orbital molecular se extiende sobre ambos átomos, los electrones de enlace deben estar asociados con ambos átomos.
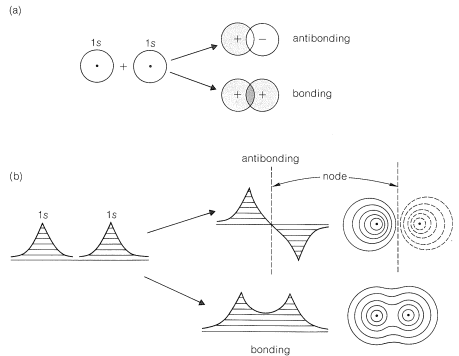
Recuerde, el método MO primero combina los orbitales atómicos para dar orbitales moleculares, luego puebla los orbitales moleculares con electrones (no más de dos electrones emparejados por orbital). Esta parte del procedimiento es similar a la forma en que se asignan los electrones a los orbitales atómicos (Sección 6-1).
El tratamiento VB comienza con los mismos orbitales atómicos pero asigna un electrón a cada orbital. Para un enlace de par de electrones entre dos átomos de hidrógeno, el tratamiento VB en su forma más simple considera dos configuraciones electrónicas. Uno de estos tiene electrón 1 en el orbital de hidrógeno 1 y electrón 2 en el orbital de hidrógeno 2,\(\left( 1 \right)\). La otra configuración,\(2\), tiene el electrón 2 en el orbital del hidrógeno 1 y el electrón 1 en el orbital del hidrógeno 2:
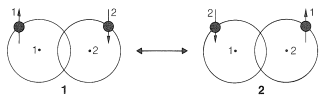
Luego, el cálculo procede a predecir un estado de baja energía y un estado de alta energía. Estos estados pueden ser considerados como híbridos de\(1\) y\(2\). El estado de baja energía, que es el que más nos interesa, suele llamarse híbrido de resonancia.
En el método VB, cada uno de los electrones se asocia con ambos átomos a través de la mezcla de las dos configuraciones. Un punto muy importante aquí es que el cálculo que mezcla\(1\) y\(2\) conduce a una energía de unión seis veces mayor que la calculada para\(1\) y\(2\) sola. Así, en el tratamiento VB combinamos configuraciones electrónicas (aquí\(1\) y\(2\),\(\leftrightarrow\) simbolizando la mezcla), mientras que en el tratamiento MO combinamos orbitales atómicos para obtener orbitales moleculares de baja y alta energía.
¿Cuál es el Pegamento en Estos Bonos?
Las fuerzas que mantienen unidos a los átomos a través de enlaces químicos son electrostáticas, es decir, la atracción de núcleos cargados positivamente para electrones cargados negativamente. Pero la energía calculada para una sola configuración, tal como\(1\), sólo representa alrededor de una sexta parte de la vinculación total. En el método VB o MO los electrones en un par de electrones entre dos núcleos llevados a distancias de unión son equivalentes e indistinguibles. Es decir, somos incapaces de identificar un electrón más que el otro con un átomo dado. La importancia del emparejamiento de los electrones es que permite que cada electrón tenga la máxima libertad posible para moverse a través de los orbitales del sistema de dos átomos en lugar de estar “localizado” en átomos particulares. Los cálculos cuántico-mecánicos nos dicen que la libertad de movimiento de los electrones es muy importante. Así, utilizando el método VB, calculamos que cinco sextas partes de la unión de la molécula de hidrógeno están asociadas con la “deslocalización” de los electrones entre los dos núcleos.
Existen muchos compuestos con estructuras en las que los electrones se deslocalizan sobre más de dos átomos. Dichas moléculas deberían ser más estables de lo que se esperaría para moléculas con la misma geometría pero con pares de electrones restringidos para asociarse con solo uno o dos átomos. En breve discutiremos algunos ejemplos específicos, pero debido a que la mayoría de estos ejemplos involucran la deslocalización de\(\pi\) electrones, es conveniente discutir primero el eteno como prototipo, utilizando los métodos MO y VB.
El\(\pi\) vínculo en el eteno
La imagen atómica-orbital del eteno (Figura 6-14) formula el\(\pi\) enlace como resultado del solapamiento de dos orbitales\(p\) atómicos adyacentes, uno de cada uno de dos carbonos\(sp^2\) hibridados. Los\(p\) orbitales se dirigen perpendicularmente al plano definido por los orbitales híbridos de los\(\sigma\) enlaces, y a una primera aproximación, asumimos que el intercambio de los\(\sigma\) electrones\(\pi\) y entre sus respectivos orbitales no afecta la energía de la molécula. Si esta suposición es válida, la\(\pi\) unión puede tratarse independientemente de la\(\sigma\) unión. Aunque indudablemente sobresimplificados, los métodos VB y MO han sido notablemente exitosos usando esta suposición. En nuestras discusiones posteriores, trataremos los\(\pi\) electrones por separado de los\(\sigma\) electrones localizados.
El\(\pi\) enlace de la molécula de eteno puede formularse muy parecido al enlace en la molécula de hidrógeno (Sección 21-2A), con la diferencia de que el enlace se logra por el solapamiento de dos orbitales\(2p\) atómicos de carbono en lugar de dos orbitales\(1s\) atómicos de hidrógeno.
En el método MO la mezcla de los dos orbitales\(2p\) atómicos da dos orbitales moleculares. Los detalles de las matemáticas del proceso de mezcla para dar un conjunto óptimo de orbitales moleculares están mucho más allá del alcance de este libro,\(^1\) pero los resultados se muestran en la Figura 21-3. Los dos\(\pi\) electrones del eteno se toman como ocupando el orbital de enlace de baja energía, mientras que el orbital antienlace de alta energía normalmente está vacío.
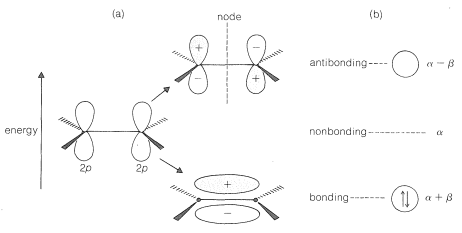
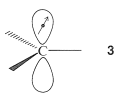
¿Cuánto más estable es el orbital molecular de unión en relación con un par de orbitales\(p\) atómicos que no interactúan? Es difícil dar una respuesta numérica en\(\text{kcal mol}^{-1}\) que sea significativa, pero podemos describir la energía en términos simbólicos. Primero, la energía de un electrón en el orbital\(p\) atómico de un\(sp^2\) carbono hibridado, como en\(3\), se toma como una cantidad estándar,\(\alpha\), a menudo llamada la energía Coulomb:
Así, si no hubiera\(\pi\) unión en eteno y no hubiera repulsión entre los electrones, la energía de los dos electrones (uno en cada uno de los dos\(p\) orbitales adyacentes de los carbonos) sería el doble de la energía de Coulomb, o\(2 \alpha\). Esta sería la situación para dos carbonos como los\(3\) que están ampliamente separados.
El cálculo de MO muestra que el orbital molecular de unión del eteno es más estable (de menor energía) que el nivel de no unión\(\alpha\), en una cantidad\(\beta\), donde\(\beta\) es un término de energía negativa (Figura 21-3). De igual manera, el nivel antiadherentes se desestabiliza en una cantidad\(-\beta\). Para dos electrones emparejados en el orbital molecular de enlace, se calcula que la energía de\(\pi\) electrones del eteno es\(2 \left( \alpha + \beta \right) = 2 \alpha + 2 \beta\).
En el enfoque de enlace de valencia, el\(\pi\) enlace de eteno se considera un híbrido de todas las configuraciones electrónicas razonables de dos electrones emparejados indistinguibles distribuidos entre dos\(p\) orbitales. Cada una de las configuraciones que se pueden escribir\(4a\),\(4b\),\(4c\), y\(4d\), tienen ubicaciones idénticas de los núcleos atómicos en el espacio:
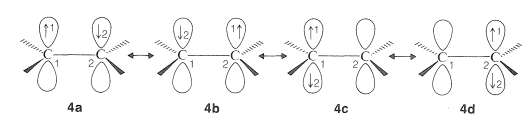
Las cuatro estructuras o configuraciones de enlace de valencia,\(4a\) -\(d\), se combinan matemáticamente para dar cuatro estados híbridos, y de estos, el de menor energía corresponde aproximadamente al estado normal de la molécula. El cálculo muestra que las estructuras\(4a\) y\(4b\), que tienen un electrón en cada\(p\) orbital, son los principales contribuyentes al “híbrido” del eteno. Las estructuras de enlace de valencia,\(4c\) y\(4d\), son estructuras iónicas, que corresponden a las fórmulas convencionales,\(4e\) y\(4f\):
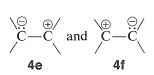
Estas estructuras de enlace de valencia no son importantes para el\(\pi\) enlace del estado fundamental del eteno, aunque son importantes para los enlaces carbonilo (Sección 16-1B).
\(^1\)Hay muchos libros excelentes que cubren este tema con gran detalle; sin embargo, el trabajo introductorio más simple es J. D. Roberts; Molecular Orbital Calculations, W. A. Benjamin, Inc., Menlo Park, Calif., 1961.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."