
21.4: El problema del benceno
- Page ID
- 72851

Ya hemos aludido a las dificultades encontradas en la interpretación de la estructura del benceno en las Secciones 1-1G y 6-5. Nuestra tarea aquí es ver qué nueva visión nos pueden dar los tratamientos VB y MO sobre el benceno, pero primero indicaremos aquellas propiedades del benceno que son difíciles de explicar sobre la base de la teoría de la estructura simple.
Algunas de las Propiedades Insólitas del Benceno
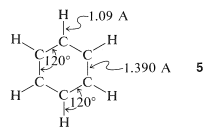
A partir de la difracción de rayos X y las mediciones espectroscópicas, se sabe que el benceno es una molécula plana con seis carbonos\(1.390\) Å separados en un anillo hexagonal,\(5\). Seis átomos de hidrógeno, uno asociado a cada carbono, se localizan\(1.09\) Å de esos carbonos. Todos\(\ce{H-C-C}\) y los ángulos de\(\ce{C-C-C}\) unión son\(120^\text{o}\):
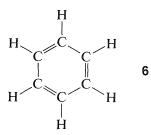
La estructura 1,3,5-ciclohexatrieno\(6\), propuesta para el benceno en 1866 por Kekule, tiene enlaces simples y dobles alternantes alrededor del anillo, lo que se predeciría que tendría longitudes de enlace de\(1.48\) Å y\(1.34\) Å, respectivamente (ver Tabla 2-1):
El conocimiento de que las longitudes de enlace son iguales en el anillo en benceno es un punto en contra de la formulación de Kekule, pero se dispone de un argumento más convincente a partir de una comparación de la química del benceno con la del 1,3,5-hexatrieno,\(7\):
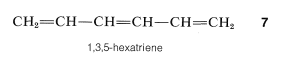
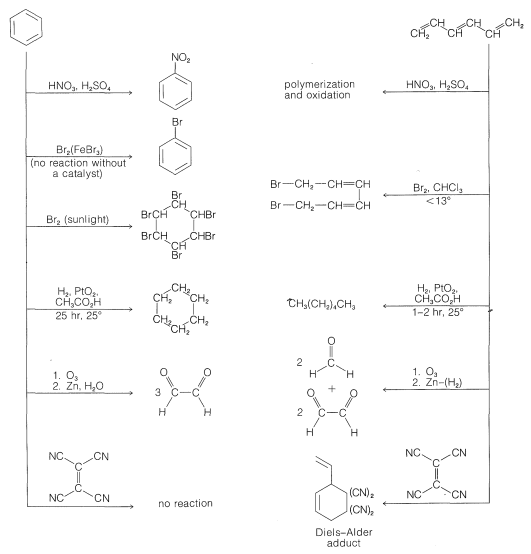
El benceno también es más estable en aproximadamente\(36\) -\(38 \: \text{kcal mol}^{-1}\) de lo previsto para la estructura 1,3,5-ciclohexatrieno. Recordará de discusiones anteriores que el calor de combustión de un mol de benceno es\(38 \: \text{kcal}\) menor que el calculado para el ciclohexatrieno (ver Sección 6-5A). Además, el calor de hidrogenación del benceno es solo\(49.8 \: \text{kcal mol}^{1}\), que es\(36 \: \text{kcal}\) menor de lo esperado para 1,3,5-ciclohexatrieno; esta estimación se basa en el supuesto de que el calor de hidrogenación de 1,3,5-ciclohexatrieno (con tres dobles enlaces) sería tres veces mayor que el del ciclohexano ( \(28.5 \: \text{kcal mol}^{-1}\), para un doble enlace), o\(3 \times 28.2 = 85.5 \: \text{kcal mol}^{-1}\).
La estabilidad extra del benceno en relación con el hipotético 1,3,5-ciclohexatrieno se puede llamar su energía de estabilización. La mayor parte (pero no toda) de esta estabilización puede atribuirse a la resonancia o deslocalización de electrones.
El modelo atómico-orbital del benceno
En la Sección 6-5 se discutió con cierto detalle un modelo atómico de benceno. Se consideró que cada carbono en el anillo formaba tres\(\sigma\) enlaces\(sp^2\) coplanares híbridos en\(120^\text{o}\) ángulos. Estos\(\sigma\) enlaces carbono-carbono y carbono-hidrógeno utilizan tres de los cuatro electrones de valencia de cada carbono. Los seis electrones de carbono restantes están en\(p\) orbitales paralelos, uno en cada uno de los seis carbonos. Cada uno de\(\pi\) los electrones puede considerarse emparejado con sus vecinos inmediatos alrededor del anillo, como se muestra por\(8\):
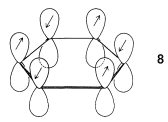
Como se menciona en la Sección 21-2B, la deslocalización de los electrones sobre los seis centros en benceno debería dar una distribución de electrones más estable que cualquier estructura en la que los electrones se localicen en pares entre carbonos adyacentes (como en la estructura clásica de 1,3,5-ciclohexatrieno).
Los tratamientos simples MO y VB del benceno comienzan con el mismo modelo atómico-orbital y cada uno trata el benceno como un problema de\(\pi\) unión de seis electrones. El supuesto es que los\(\sigma\) enlaces del benceno no deben ser muy diferentes de los del eteno y pueden considerarse como independientes del\(\pi\) sistema.
El método orbital molecular para el benceno
La extensión de las ideas de la Sección 21-2 para el tratamiento MO de un enlace de par de electrones entre dos núcleos y el\(\pi\) enlace en benceno es bastante sencilla. Lo que es muy importante entender es que debe haber más de un orbital molecular para los\(\pi\) electrones porque hay seis\(\pi\) electrones, y el principio Pauli no permite que más de dos electrones pareados ocupen un orbital dado. De hecho, la combinación (o mezcla) de los seis\(2p\) orbitales de benceno, mostrados en\(8\), da seis orbitales\(\pi\) moleculares. Sin excepción, el número de orbitales moleculares obtenidos por mezcla es siempre el mismo que el número de orbitales atómicos mezclados. Aquí no se describirán los detalles de las matemáticas del proceso de mezcla para dar un conjunto óptimo de orbitales moleculares,\(^1\) pero los resultados se muestran en la Figura 21-5. De los seis orbitales moleculares previstos, tres son enlazantes y tres son antiadherentes. Los seis\(\pi\) electrones se asignan a los tres orbitales de enlace en pares y se calculan para tener una energía total\(\pi\) de electrones de\(6 \alpha + 8 \beta\).
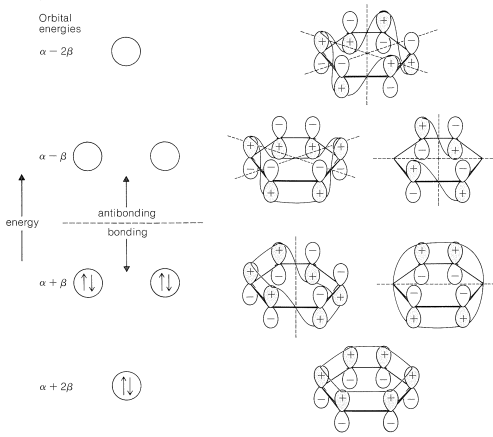
El cálculo que lleva a los resultados mostrados en la Figura 21-5 no es muy sofisticado. Se basa en la suposición de que la\(\pi\) unión entre cada carbono y sus vecinos inmediatos es igual alrededor del anillo y que la unión que involucra carbonos más de\(2\) Å separados no es importante.
¿Qué sucede si utilizamos el método MO para calcular la energía\(\pi\) -electrónica del 1,3,5-ciclohexatrieno clásico? El procedimiento es exactamente como para el benceno, salvo que decretamos que cada\(p\) orbital de carbono forma un\(\pi\) enlace con solo uno de sus\(p\) orbitales vecinos. Los resultados se muestran en la Figura 21-6. La energía\(\pi\) -electrónica resulta ser tres veces mayor que la del eteno, o\(6 \alpha + 6 \beta\) comparada con la\(6 \alpha + 8 \beta\) del benceno.
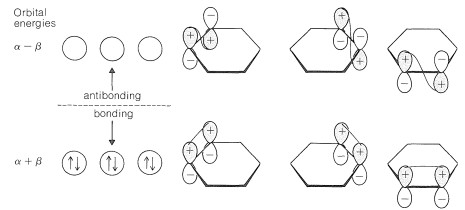
La energía de deslocalización calculada para el benceno es la diferencia entre estas cantidades, o\(\left( 6 \alpha + 8 \beta \right) - \left( 6 \alpha + 6 \beta \right) = 2 \beta\). Es decir, la energía de deslocalización calculada es la diferencia entre la energía del benceno con\(\pi\) enlace completo y la energía del 1,3,5-ciclohexatrieno con enlaces simples y dobles alternantes. Si la energía de deslocalización de electrones\(\left( 2 \beta \right)\) es igual a la energía de estabilización\(\left( 38 \: \text{kcal mol}^{-1} \right)\), entonces\(\beta = 19 \: \text{kcal mol}^{-1}\). El hecho de que se trate de un método válido para determinar\(\beta\) ha sido objeto de controversia desde hace muchos años. Independientemente de esto, los resultados de los cálculos sí explican el hecho de que el benceno es más estable de lo que se esperaría para 1,3,5-ciclohexatrieno.
Sin embargo, ¿los resultados también explican la baja reactividad hacia los diversos reactivos de la Figura 214, como los que donan\(\ce{Br}^\oplus\) a dobles enlaces (ver Sección 10-3A)? Para resolver esta cuestión, hemos calculado los cambios en la energía\(\pi\) electrónica que ocurren en cada una de las siguientes reacciones:
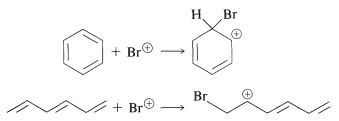
Esto significa calcular las energías\(\pi\) de electrones de las cuatro entidades y asumir que las diferencias en las energías\(\sigma\) de enlace se cancelan entre las dos reacciones. El resultado de este cálculo bastante simple es que\(\ce{Br}^\oplus\) el ataque de benceno es termodinámicamente menos favorable que sobre 1,3,5-ciclohexatrieno en aproximadamente\(\beta\). Si\(\beta\) es así\(19 \: \text{kcal mol}^{-1}\), esta es claramente una diferencia de energía considerable, y podemos concluir que el método simple MO realmente explica el hecho de que el benceno es atacado con\(\ce{Br}^\oplus\) mucha menos facilidad que el 1,3,5-ciclohexatrieno.
El método de enlace de valencia para benceno
La extensión de las ideas básicas del tratamiento VB descritas en la Sección 21-2 al modelo atómico-orbital del benceno es sencilla. Podemos escribir estructuras VB que representan esquemas de emparejamiento de electrones en los orbitales atómicos como se muestra en\(9\) a través de\(13\):
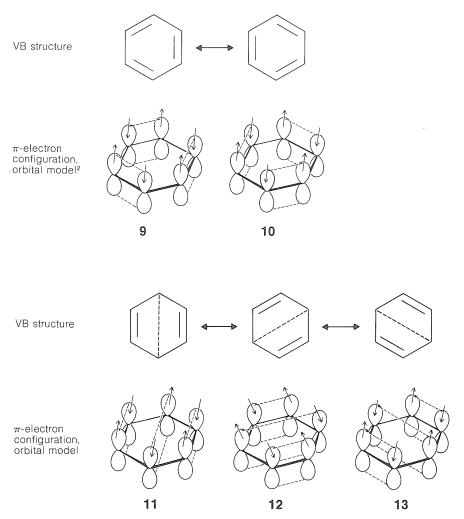
Los esquemas de emparejamiento\(9\) y\(10\) corresponden a las estructuras de Kekule\(11\)\(12\), mientras que, y\(13\) se llaman “estructuras Dewar” porque J. Dewar sugirió, en 1869, que el benceno podría tener una estructura como\(14\):
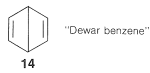
Los electrones están emparejados en las configuraciones representadas por\(11\),\(12\), y\(13\), pero estos esquemas de emparejamiento no son tan energéticamente favorables como\(9\) y\(10\). La razón es que los dos electrones se emparejaron de acuerdo con las líneas discontinuas en\(11\)\(12\),, y\(13\) están en núcleos separados por\(2.8\) Å, que está demasiado lejos para una unión efectiva. Las líneas discontinuas entre los carbonos distantes en\(11\)\(12\),, y\(13\) son significativas solo en que definen un esquema de emparejamiento. A veces se dice que tales líneas representan “bonos formales”.
Esperamos que quede claro por lo que hemos dicho aquí y anteriormente que los esquemas de apareamiento de electrones\(9\) a través\(13\) no tienen por separado la realidad física o la existencia independiente; en efecto, la energía de la molécula real es menor que cualquiera de los contribuyentes estructuras. La flecha de doble punta entre las estructuras se utiliza para indicar que representan diferentes esquemas de emparejamiento de electrones para una molécula y no diferentes formas de la molécula en equilibrio entre sí. Cuando utilizamos el método de resonancia de manera cualitativa, consideramos que la contribución de cada una de las diversas estructuras se va a ponderar de alguna manera que concuerde con el grado de unión que cada una tendría, si fuera a representar una molécula real con la geometría especificada. Así, los esquemas de apareamiento de electrones tipo Kekule-type\(10\),\(9\) y, deben tomarse como contribuyentes igual y predominantemente a la estructura híbrida del benceno -igualmente porque son energéticamente equivalentes, y predominantemente porque pueden contribuir mucho más a la unión general que\(11\),\(12\), y\(13\).
Al usar el método de resonancia, asumimos que todas las estructuras de resonancia que contribuyen a un híbrido de resonancia dado tienen exactamente las mismas disposiciones espaciales de los núcleos pero diferentes esquemas de emparejamiento para los electrones. Por lo tanto\(11\)\(12\),, y\(13\) no deben confundirse con biciclo [2.2.0] -2,5-hexadieno,\(15\), debido a que\(15\) es una molécula conocida (aunque no muy estable) con diferentes posiciones de átomo y por lo tanto ángulos de enlace muy diferentes y longitudes de enlace del benceno:
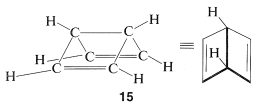
Los esquemas de apareamiento de electrones\(9\) y\(10\) representan el apareamiento electrónico que\(15\) tendría si estuviera muy distorsionado, con cada carbono en la esquina de un hexágono regular y un enlace formal en lugar de un enlace sencillo carbono-carbono. Así\(9\) y no\(10\) contribuiría de manera significativa al híbrido de resonancia de\(15\).
Claramente, es inconveniente y tedioso escribir las estructuras de las formas contribuyentes para mostrar la estructura de un híbrido de resonancia. Por lo tanto, es deseable una notación taquigráfica. Frecuentemente, se utilizan líneas discontinuas en lugar de líneas completas donde se espera que los electrones de enlace se deslocalicen sobre varios átomos. Para benceno,\(16a\) o\(16b\) es bastante apropiado:

Desafortunadamente, aunque se trata de representaciones claras y explícitas, son tediosas de dibujar. Como resultado, muchos autores utilizan (como lo haremos con mayor frecuencia) una sola estructura Kekule para representar el benceno, entendiendo que todos los\(\ce{C-C}\) enlaces son equivalentes. Otros autores optan por representar el benceno como un hexágono con un círculo inscrito:
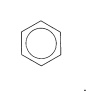
Esta es una notación simple para el benceno, pero es bastante poco informativa e incluso puede ser engañosa activamente con algunos sistemas de anillos aromáticos, y por lo tanto debe usarse con esta limitación en mente.
En los cálculos de la energía de resonancia del benceno, las cinco configuraciones electrónicas (estructuras\(9\) de enlace de valencia\(13\)) se combinan matemáticamente para dar cinco estados híbridos, y de estos se supone que el estado de menor energía corresponde al estado normal de la molécula. Por lo tanto, el benceno es considerado por este enfoque como un híbrido de resonancia de las estructuras de enlace de valencia\(9\) a través de\(13\). En este tratamiento sencillo,\(9\) y\(10\) se calculan para contribuir sobre\(80\%\) y\(11\),\(12\), y\(20\%\) a\(13\) punto del híbrido. Los cálculos numéricos reales de VB, que son mucho más difíciles de llevar a cabo que los correspondientes cálculos MO, dan una energía de\(Q + 2.61 J\) para benceno y\(Q + 1.50 J\) para 1,3,5-ciclohexatrieno clásico. \(^3\)La energía de resonancia o deslocalización entonces es\(\left( Q + 2.61 J \right) - \left( Q + 1.50 J \right) = 1.11 J\), lo que hace que\(J \sim 35 \: \text{kcal mol}^{-1}\) si se toma la energía de resonancia sea igual al\(38 \: \text{kcal}\) valor obtenido para la energía de estabilización. Si se lleva a cabo un simple cálculo VB del cambio de energía relativa asociado con el ataque de benceno\(\ce{Br}^\oplus\) en comparación con 1,3,5-ciclohexatrieno, el valor obtenido es\(0.63 J\), que corresponde a\(22 \: \text{kcal}\). Esto está en excelente acuerdo con el\(19 \: \text{kcal}\) valor obtenido por el método MO (Sección 21-3C).
\(^1\)Hay muchos libros excelentes que cubren este tema con gran detalle; sin embargo, el trabajo introductorio más simple es J. D. Roberts, Molecular Orbital Calculations, W. A. Benjamin, Inc., Menlo Park, Calif., 1961.
\(^2\)Tenga en cuenta que en\(9\) y también en\(10\), mostramos una forma particular de emparejar los electrones. Sin embargo, así como\(1 \leftrightarrow 2\), y\(4a \leftrightarrow 4b\), también debemos considerar otros conjuntos que representan intercambios de electrones a través de las líneas discontinuas de\(9\) y también de\(10\).
\(^3\)\(Q\)y\(J\) son parámetros de energía VB negativos que corresponden aproximadamente a los parámetros MO\(\alpha\) y\(\beta\).
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."