
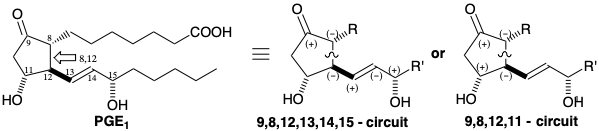
3.3: Síntesis de Prostaglandinas a partir de Precursores Acíclicos
- Page ID
- 70163

Al igual que en la biosíntesis de prostaglandinas, varias síntesis totales presentan la generación del anillo de ciclopentano por ciclación de precursores acíclicos. Primero compararemos la biosíntesis con tres síntesis totales que implican la formación del anillo de ciclopentano por generación del enlace de prostaglandina 8,12. Dado que el enlace 8,12 se encuentra en circuitos disonantes entre la funcionalidad en las posiciones 9 y 11 o en las posiciones 9 y 15, no se puede lograr una conexión polar que depende de la activación polar por cualquiera de los dos pares de grupos funcionales. Por lo tanto, cada una de estas síntesis explota la activación polar proporcionada por un solo grupo funcional relacionado con la diana para promover la creación polar del enlace 8,12. En la estrategia 1 de Miyano (ver abajo), la desconexión polar de la cadena lateral inferior de PGE 1 en el enlace C=C sugiere la subdiana 19. Ni el grupo aldehído ni la funcionalidad en C-11 en 19 pueden proporcionar la electrofilicidad requerida en C-12 para la formación de enlaces polares aprovechando la nucleofilia en C-8 activada por el carbonilo C-9. Por lo tanto, un precursor de 19 debe incorporar funcionalidad adicional para proporcionar electrofilicidad en el C-12 incipiente. Esto podría ser proporcionado por un grupo carbonilo como en 20. Sin embargo, esta estrategia es defectuosa ya que el aldehído electrófilo en 20 competirá con el carbonilo en el incipiente C-12 en 20 por la reacción con un nucleófilo C-8. Esto producirá un subproducto de benzoquinona no deseado derivado del intermedio 21 por deshidratación.
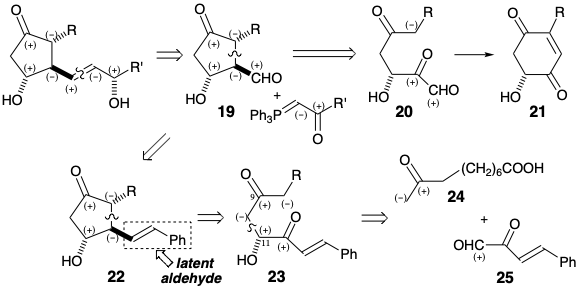
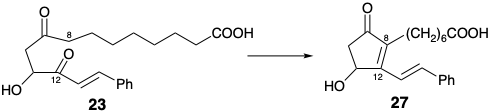
Para evitar la competencia indeseada por un aldehído electrófilo, este grupo no se introduce hasta que es necesario. En cambio, se oculta en un precursor 22 de 19 como un enlace C=C a partir del cual se puede generar por escisión oxidativa. El enlace C=C no posee la alta electrofilicidad de un aldehído. Sin embargo, se puede generar fácilmente un grupo aldehído a partir del enlace C=C. La desconexión polar de 22 sugiere funcionalidad electrofílica en la prostaglandina C-12 incipiente en un precursor 23. La desconexión adicional de 23 a 24 y 25 explota la activación polar proporcionada por la funcionalidad consonante en los incipientes C-9 y C-11 ignorando el carbonilo en C-12 en 23.
Nos referiremos a un grupo precursor directo no reactivo con un nivel de funcionalidad diferente al objetivo como grupo funcional latente (ver a continuación para algunos ejemplos). 2 Así, (1) un alqueno (f = 0), como en 22 anterior, o un diol vecinal (f = 1) pueden servir como equivalente latente de un aldehído o cetona (f = 2). Si bien el alcohol es electrófilo en el carbono carbonílico incipiente, su electrofilicidad es considerablemente menor que la de un aldehído o cetona. (2) Un areno (f = 0) es un grupo carboxilo latente (f = 3). (3) Un enol éter (f = 1) proporciona un éster latente (f = 3) ya que se puede lograr la escisión oxidativa del precursor latente para proporcionar el grupo funcional deseado. (4) Una cetona (f = 2) es un éster latente (f = 3) ya que la oxidación de Baeyer-Villiger del primero entregará la última; y (5) un alquino terminal (f = 0) es un anión acetiluro latente (f = -1) o carbanión vinílico terminal (f = -1) equivalente ya que el hidrógeno alquinílico se abstrae fácilmente por bases fuertes y los productos alquinos del acoplamiento de acetiluros con electrófilos pueden reducirse selectivamente al alqueno correspondiente. Nótese que la generación de un grupo funcional a partir de su equivalente latente, por definición, implica oxidación [O] o reducción [R], es decir, aumento o disminución del nivel de funcionalidad. También tenga en cuenta que la generación de un carbanión por abstracción de protones a partir de la adición de carbono o hidruro a un compuesto de carbonilo α, β- insaturado corresponde a una reducción. En otras palabras, la abstracción de protones a partir de un “ácido carbonado”, es decir, un enlace C-H ácido, corresponde a la reducción en el nivel de funcionalidad del carbono del que se extrae un protón y la oxidación del hidrógeno que se abstrae como protón. También tenga en cuenta que la adición de un protón a un enlace C=C da como resultado la reducción del protón y oxidación del carbono que se convierte en carbocatión, y la adición de\(\ce{Cl2}\) a un enlace C=C corresponde a la oxidación de ambos carbonos con oxidación concomitante de ambos cloros, proceso que se denomina “dioxidativo adición”.
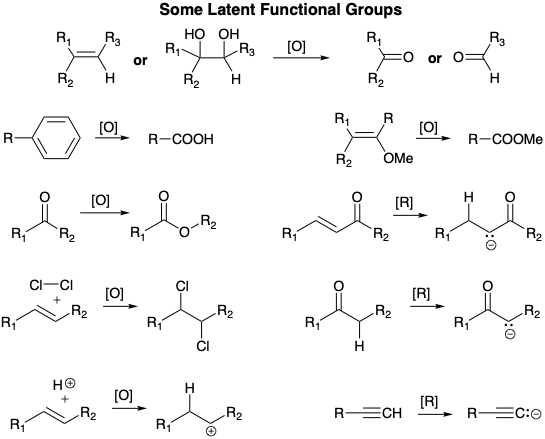
Un concepto estrechamente relacionado es el grupo funcional enmascarado, que es un grupo precursor no reactivo con el mismo nivel de funcionalidad que el objetivo. Por ejemplo, un cetal es una cetona enmascarada (f = 2). Otro ejemplo lo proporcionan los ésteres (f = 3) que sirven como grupos carboxilo enmascarados (f = 3). La esterificación bloquea la proclividad del ácido hacia la descarboxilación cuando β a un carbonilo o hacia la desprotonación por bases. Como veremos en la síntesis de Kojima-Saki de prostaglandinas (ver más adelante), diferentes grupos enmascarantes para un mismo grupo funcional, como un bencilo y un éster metílico para dos grupos ácido carboxílico en una molécula, pueden permitir la eliminación selectiva (desenmascaramiento) para convertir un éster en un ácido mientras que el otro permanece enmascarado.
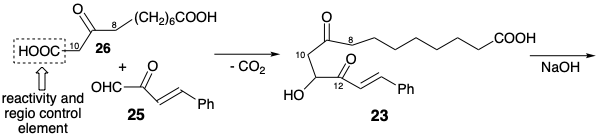
En la síntesis de Miyano de PGE 1, el β-cetoácido 26 en lugar de la metil cetona 24 (ver arriba) se condensa con cetoaldehído 25. Esta condensación descarboxilativa logra regioselectivamente la activación nucleofílica en la incipiente posición 10 sin competencia por la condensación en la incipiente posición 8 que también puede ser activada por el carbonilo C-9. Así, el grupo carboxilo unido a la posición 10 en 26 sirve como grupo activador. Se trata de un elemento de control de la reactividad y, en consecuencia, de un elemento de regiocontrol. La condensación intramolecular de aldol genera entonces el enlace 8,12 que suministra ciclopentenona 27. La escisión oxidativa selectiva del enlace estirilo C=C más rico en electrones en 27 proporciona un aldehído insaturado 28. La saturación del enlace C=C restante proporciona 29 en los que los grupos aldehído y carboxiheptilo adoptan las configuraciones relativas trans requeridas debido a una preferencia termodinámica y epimerizabilidad en ambas posiciones 8 y 12. La condensación de 29 con iluro 30 luego entrega PGE1 después de la reducción selectiva del 15-carbonilo. Ni el estereocentro en la posición 15 ni el de la posición 11 se genera con alto estereocontrol: control termodinámico (TC). Además, esta síntesis produce una mezcla racémica de PGE 1 y su enantiómero no natural ya que cualquier síntesis que comience con materiales de partida no asimétricos o racémicos y emplee reactivos racémicos finalmente generará solo productos racémicos.
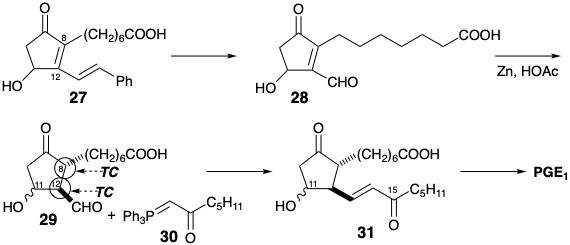
Una segunda estrategia 3 para la síntesis de prostaglandinas que implica la anulación de ciclopentan-ona mediante la formación del enlace 8,12 comienza con la desconexión polar de la cadena lateral inferior de PGF2α para sugerir una subdiana 32. Sin embargo, en lugar de utilizar un nucleófilo C-8 y un electrófilo C-12 como en la síntesis de Miyano, la síntesis de Kojima-Saki logra la formación de un enlace polar 8,12 por reacción de un electrófilo C-8 con un nucleófilo C-12. El aldehído C-12 en 32 o un éster en un precursor 33 pueden activar la generación de carbaniones en C-12. Para impedir la β-eliminación del oxígeno C-ll y proporcionar una estabilización adicional de un carbanión C-12, el oxígeno C-11 está presente como grupo carbonilo en el precursor 33 de 32. Una posible β- eliminación del oxígeno C-9 se excluye al ocultar este grupo funcional en forma latente como un grupo benciloxicarbonilo en 33. La desconexión polar de 33 requiere reactividad electrófila en C-8 que podría ser proporcionada por conjugación con el benciloxicarbonil o un grupo carbonilo adicional en C-8 como en 34. Este grupo carbonilo también puede proporcionar activación nucleofílica en C-9 para la generación polar del enlace 9,10. El benciloxicarbonil también puede ayudar en la formación de un carbanión en C-9 y prevenir la interferencia por la generación de carbaniones en C-7. Así, el benciloxi-carbonilo también sirve como un grupo activador y un elemento regiocontrol. El circuito 9,10,11 en 34 es disonante. Por lo tanto, la desconexión polar en un C-9 nucleofílico requiere la activación electrofílica del C-10 incipiente en un precursor 36. El carbonilo en C-11 en 34 se ignora en la desconexión polar a 36 que es un sintón umpoled del acetoacetato de metilo (37).
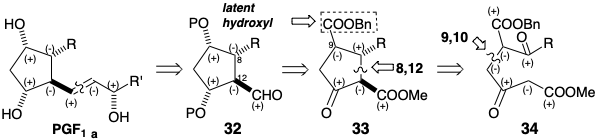
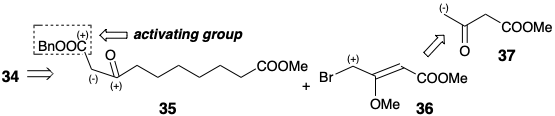
Al igual que en la ciclación 23 a 27 de la síntesis de Miyano, la ciclación de 34 produce una ciclopentanona 38. Pero ahora se invierten los papeles de nucleófilo y electrófilo. Así, en la síntesis de Miyano el centro nucleofílico está en la posición 8 y el centro electrófilo en la posición 12 en 23.
En la síntesis de Kojima-Saki, el centro nucleofílico está en la posición 12 y el centro electrófilo en la posición 8 en 34. Este intermedio se convierte estereoselectivamente en 33 por hidrogenación catalítica que satura el enlace C=C y también convierte selectivamente el éster bencílico en un ácido carboxílico sin afectar a los ésteres metílicos o etílicos. Generación preferencial de las configuraciones relativas α, α, β en C-9, 8 y 12 respectivamente en 33 resultados del control de abordaje estérico seguido de epimerización del grupo β-cetocarbometoxilo. La conversión de 33 en PGF 1α es sencilla.
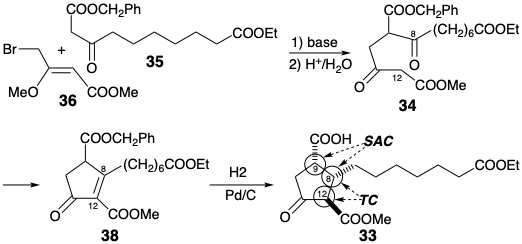
La generación del hidroxilo C-10 a partir del precursor latente, un benciloxicarbonilo C-10, se logra mediante la oxidación de Baeyer-Villiger de una metil cetona intermedia 39. La reducción selectiva del carboxilo en 39 en C-12 se logra enmascarando la reactividad electrófila del carboxilo C-1 como una sal de potasio. Para permitir la manipulación selectiva del alcohol resultante, los hidroxilos en las posiciones 9 y 11 en 40 se enmascaran como éteres de tetrahidropiranilo (THP) antes de la reducción del grupo carbometoxi.
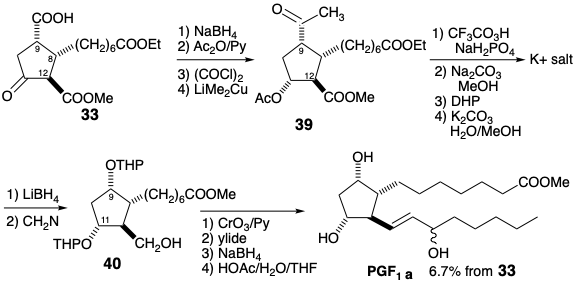
Para una síntesis de PGE 1 a partir de 39, la generación de la funcionalidad oxígeno C-9 a partir de su precursor latente se retrasa hasta después de que el grupo acetoxi C-11 en 39 se convierta en un éter THP para permitir la diferenciación del acetoxi C-9 generado en una oxidación Baeyer-Villager de 43. El grupo protector ditiocetal en 41 se puede eliminar selectivamente de 42 después de la introducción del éter de THP. La adición de la cadena lateral inferior a 43 después de la oxidación al aldehído 44 es paralela a la secuencia descrita para la síntesis de Miyano.
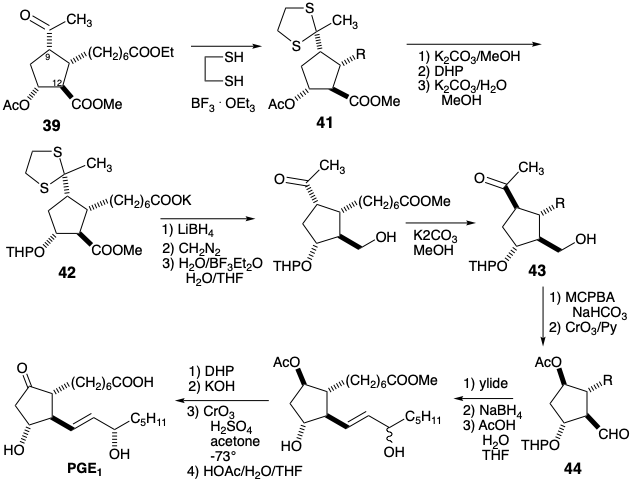
La matriz β-hidroxicarbonil en PGE 2 es sensible a la deshidratación. Si se va a explotar el carbonilo C-9 para activar la reactividad nucleofílica en C-8 en un precursor, esta sensibilidad debe ser abordada. En la síntesis de Miyano, la deshidratación del producto de ciclación 27 se ve desfavorecida por la inestabilidad de la ciclopentadienona antiaromática que resultaría. En una tercera estrategia para la síntesis de prostaglandinas4 que también imita la anulación biosintética de ciclopentano que implica la creación de enlaces 8,12, el problema se elude introduciendo el hidroxilo C-11 al final de la síntesis. Esto sugiere un intermedio clave 45 en el que se elimina la funcionalidad C-11 y se agrega un grupo activador en C-8 para ayudar en la generación de carbaniones en esta posición. La desconexión polar de la cadena lateral superior de la subdiana 45 sugiere una funcionalidad electrofílica en C-7 como en el precursor 50. La desconexión polar de 46 en el enlace 8,12- revela la necesidad de electrofilicidad en C-12. Sin embargo, en lugar de colocar la funcionalidad apropiada en C-12 como en el intermedio 23 de la síntesis de Miyano (ver arriba), un nucleófugo en C-14 en 47 proporciona la activación requerida por conjugación con C-12. Un epóxido 47a proporciona la electrofilicidad apropiada en C-12 y funcionalidad de oxígeno en C-15. Una clorohidrina 47b es un equivalente sintético alternativo que es interconvertible con 47a. El análisis polar de 47b sugiere la desconexión del precursor electrófilo 49 y 48 cuyo acetileno terminal sirve como nucleófilo vinil latente. La utilidad de los acetilenos terminales como nucleófilos vinílicos terminales latentes también sugiere una estrategia para una ruta conectiva C-C a 50 vía 51 de 52, cianuro y un dielectrófilo de 1,3-propano. Aunque 47a y 47b son equivalentes sintéticos alternativos, la conversión de 47b a 47a durante la síntesis proporcionaría un electrófilo más reactivo.
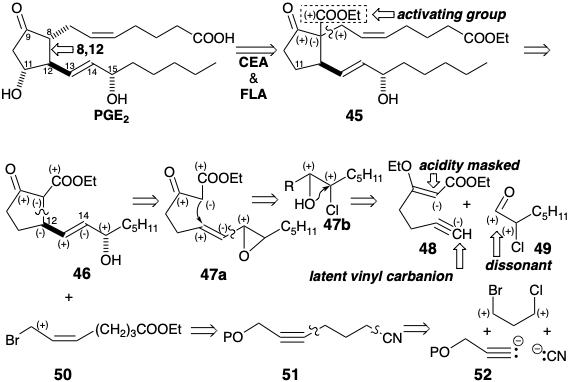
Se podría haber logrado una síntesis de 48 por propargilación del dianión etilacetoacetato. Sin embargo, el potencial de abstracción del hidrógeno acetilénico, por el dianión fuertemente básico, recomendó un enfoque alternativo.
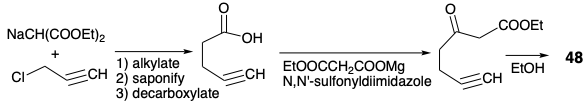
Así, una síntesis de éster malónico proporcionó ácido 4-pentinoico que fue posteriormente elaborado a un β-cetoéster por condensación de Claisen. 4 La enoleterificación luego enmascara la cetona en 48 y permite la desprotonación selectiva en el acetileno terminal. La condensación del nucleófilo acetiluro resultante con α-cloroheptanal (49) produce 53 estereoselectivamente y luego cis vinil trans epóxido 47a después de hidrogenación parcial y heterociclización promovida por bases.
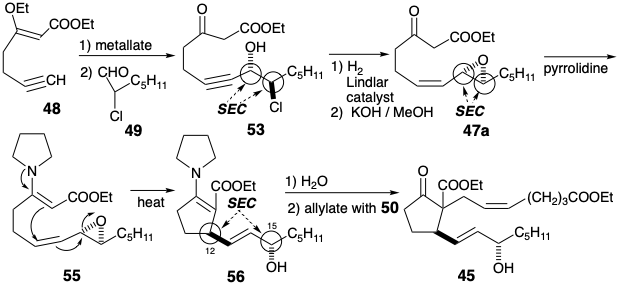
La ciclación del β-ceto-éster 47, generando el enlace de prostaglandina 8,12, podría haberse logrado mediante la alquilación intramolecular de un enolato intermedio. Sin embargo, se empleó un nucleófilo alternativo, la enamina 55. Hidrólisis de un derivado intermedio de enamina 56 seguido de alilación del β-cetoéster resultante entregado 45. Destaca especialmente la estereoselectividad de esta síntesis de 45 con la estereoquímica relativa correcta en C-12 y C-15. Esta es la consecuencia de tres reacciones consecutivas controladas estereoelectrónicamente (SEC).
Primero, la adición de un nucleófilo acetiluro derivado de 48 al α-cloroaldehído 49 ocurre estereoselectivamente en la cara menos impedida del grupo carbonilo en una conformación de 49 que logra la máxima separación de los dipolos C=O y C-Cl por un anti periplanar arreglo. La posterior ciclación del β-cloroalcóxido 54 con inversión Walden produce un epóxido trans disustituido estereoespecíficamente.
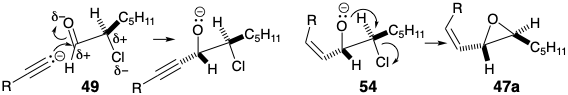
Se esperaba que un modo de ciclación estereoelectrónicamente preferido tradujera la relación estereoquímica entre los centros quirales en el epóxido cis vinil trans 55 en la relación estereoquímica requerida entre los centros quirales en las posiciones 12 y 15 en 56. 4 Así, se esperaba que el desplazamiento anti S N 2' del alcóxido por el nucleófilo enamina en 55 generara 56 después de la transferencia de protones en un supuesto intermedio de alcóxido de iminio.
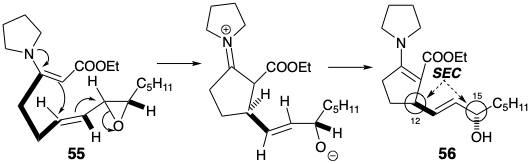
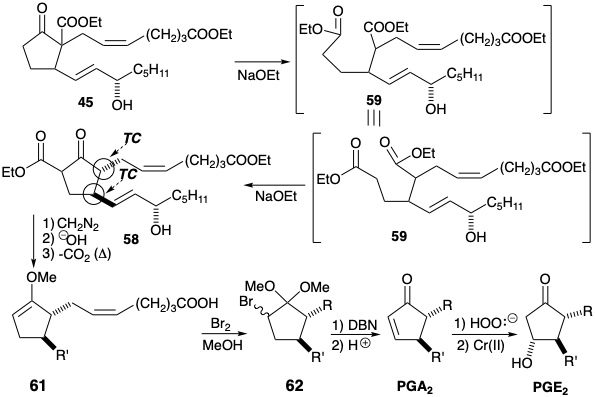
La introducción de un sustituyente hidroxilo en la posición 11 en 45 se complica por la ausencia de funcionalidad activadora adyacente a C-11. La reactividad se puede proporcionar introduciendo insaturación entre los carbonos 10 y 11 aprovechando el carbonilo C-9 para activar C-10 para introducir un grupo saliente. La PGE 2 podría entonces obtenerse por epoxidación de la cetona α, β-insaturada PGA 2 seguida de escisión reductora del enlace C-O adyacente al carbonilo en 57. Esto sugiere PGA 2 como precursor sintético de PGE 2.
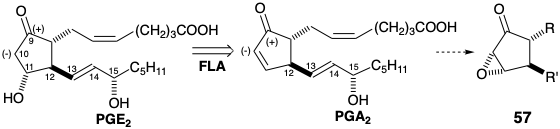
Mientras que 45 (ver arriba) podrían convertirse en PGA 2 por descarbetoxilación de la matriz β-cetoéster y posterior introducción oxidativa de un grupo saliente en C-10, se adoptó una estrategia más elaborada. El proceso proporciona un ejemplo de una construcción del esqueleto de prostaglandina al generar el enlace 9,10 del anillo de ciclopentano como la última conexión esquelética. Así, un precursor de 10-carboetoxi 58 sería muy adecuado para la activación nucleofílica regioselectiva (α vs α') (abstracción de protones) de la posición 10 requerida para la conversión de un derivado 10,11-dihidro-PGA 2 en PGA 2. Además, 58 se funcionaliza adecuadamente para la generación polar por reacción de un nucleófilo C-10 con un centro electrófilo C-9 en un precursor 59. La reconexión polar de 59 sugiere el intermedio clave 45 como precursor de 58 vía 59.
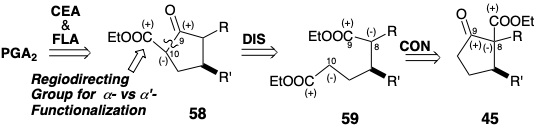
La PGA 2 es un producto de deshidratación de origen natural a partir de PGE 2, y el primero se ha preparado a partir de este último mediante una secuencia de reducción de oxidación. La estereoselectividad durante la epoxidación se logró empleando un grupo bloqueante estéricamente exigente unido al 15-hidroxilo en 60 para dirigir la epoxidación a la cara α del anillo de ciclopentenona. 5
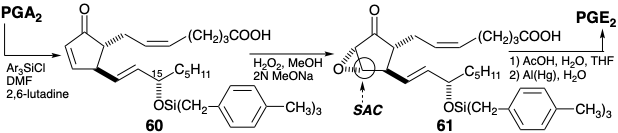
La conversión de 45 en 58 se logró mediante un reordenamiento catalizado por etóxido presumiblemente involucrando escisión retro de Dieckmann a 59 seguido de ciclación de Dieckman a 58. La transferencia de carboetoxi también puede ocurrir por descarboetoxilación y recarboetoxilación. En todo caso, el reordenamiento es impulsado a 58 por la formación del enolado correspondiente. El reordenamiento permite regiocontrol en la formación de un enol éter 61, la posterior introducción de bromo por adición 1,2-dioxidativa para dar 62, y finalmente en la introducción de insaturación entre los carbonos 10 y 11.
Los tres ejemplos anteriores de síntesis de prostaglandinas ilustraron la síntesis polar del enlace 2,3 de un anillo de ciclopentanona correspondiente al enlace 8,12 del esqueleto de prostaglandina. En cada caso, esto requería funcionalización adicional de un precursor debido a que los circuitos 9,8,12,13,14,15 o 9,8,12,11-son disonantes. Dado que el circuito 9,10,11 es consonante, el enlace 9,10 o 10,11- puede estar formado por una reacción polar que depende únicamente de la funcionalidad relacionada con la diana. Como veremos en la sección 3.7, las prostaglandinas secas (moléculas que carecen de un enlace C-C del esqueleto de la prostaglandina) son productos naturales para los que se acuñó el nombre levuglandina (LG) para significar que son derivados de levulinaldehído con cadenas laterales de prostaglandina. En teoría, 13,14-dihidro-PGE 1 (63) podría generarse directamente a partir de dihidro-LGE 1 por reacción polar de un enolato nucleofílico C-10 con el carbono carbonílico electrófilo del grupo aldehído en C-11, es decir, una condensación aldólica. Sin embargo, aunque 11,13-dihidro-PGE 1 es indudablemente un intermedio en esta ciclación, no es estable y se deshidrata bajo las condiciones básicas de la reacción de condensación aldólica para proporcionar la olefina 64, que luego debe ser refuncionalizada para suministrar la β-hidroxicetona objetivo 63 . 6
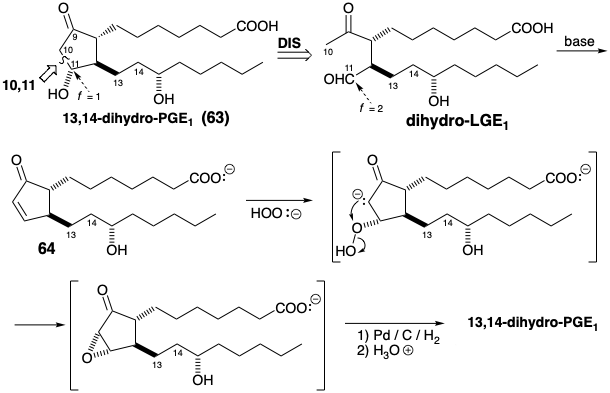
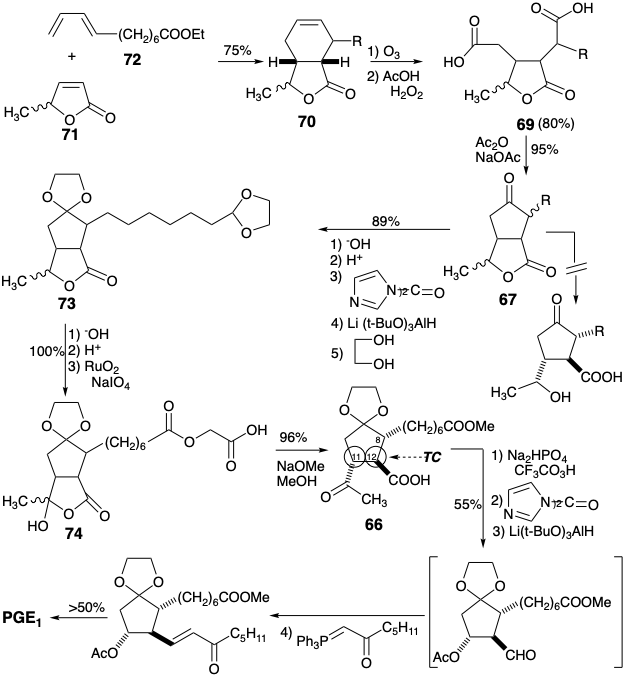
Una síntesis 7 de Merck de PGE1 logra la anulación de ciclopentanona mediante la creación del enlace 9,10 en un proceso polar. Sin embargo, para evitar la generación de un intermedio β-hidroxicetona sensible, solo se explota un grupo funcional relacionado con la diana. La desconexión polar de la cadena lateral inferior sugiere un precursor 65 (ver más abajo). El hidroxilo C-11 se oculta en forma latente hasta el final de la síntesis para evitar la β-eliminación. Así, la subdiana 65 se disloca a un penúltimo precursor 66 en el que el hidroxilo C-11 se reemplaza por una metil cetona y el carbonilo C-9 se enmascara para permitir la oxidación selectiva de Baeyer-Villiger de la metil cetona para suministrar 65. La funcionalidad reactiva de metil cetona y ácido carboxílico en 66 puede enmascararse internamente como lactona en un precursor 67. Aunque la unión del anillo en 67 es necesariamente cis, los sustituyentes en ambas posiciones 11 y 12 en 66 y sus estereoisómeros son epimerizables permitiendo la generación de la configuración relativa todo-trans requerida de los sustituyentes en 66 que se favorece termodinámicamente.

Para facilitar la síntesis polar del anillo de ciclopentanona en 67, se puede agregar un grupo carboxilo al incipiente C-10 en un precursor 69 para proporcionar activación nucleofílica (CEA). Este carboxilo activante se eliminará fácilmente del producto de ciclación 68 mediante una escisión polar. Los dos grupos ácido carboxílico reactivos en 69 pueden derivarse de un precursor latente 70 por escisión oxidativa. Finalmente, el resto ciclohexeno en 70 sugiere una doble desconexión a 71 y 72 que proporcionaría el anillo de ciclohexeno mediante una cicloadición de Diels-Alder. Se predice una preferencia por la orientación requerida de la cicloadición debido a los efectos de superposición orbital que favorecen una relación orto entre el sustituyente en el dieno y el sustituyente extractor de electrones en el dienófilo.
Durante la síntesis, se descubrió que no se pudo lograr la apertura de la lactona en 67, para permitir la oxidación del hidroxilo secundario y entregar la cetona 66. Para sortear esta falla en el plan original, 67 se convirtió en un acetal 73 que luego podría convertirse en un hemiacilo 74 por\(\ce{RuO4}\) oxidación. La transesterificación reemplazó el éster carboximetílico en 74 por un éster metílico en 66 que luego podría convertirse en PGE 1 según lo planeado.
En la síntesis anterior, se aplicó una cicloadición para generar un ciclohexeno mediante un proceso de doble conectivo C-C. Las cicloadiciones generalmente proporcionan una ruta no polar valiosa a diversos productos cíclicos. La funcionalidad es innecesaria para las cicloadiciones. Por lo general las dianas contienen instauración, pero los ciclobutanos, que pueden generarse por cicloadición, ni siquiera tienen instauración. Una dislocación retro cicloadición (RC) es factible si un desplazamiento cíclico de dos enlaces σ y n-2 enlaces π escinde el anillo en dos precursores de poli y/o monoeno. Las dianas que son candidatas para la síntesis por cicloadición de dos poli y/o monoenos pueden reconocerse por la presencia de al menos n-2 enlaces π en un circuito cerrado de 2n átomos. Si los precursores están puenteados, la cicloadición será intramolecular. Las cicloadiciones pueden ocurrir fácilmente con activación térmica si n es un número impar pero pueden requerir activación fotoquímica o catálisis de metales de transición si n es un número par. Estas reglas son consecuencia del requisito de superposición positiva entre el orbital molecular desocupado (MO) más bajo de un reactivo con el orbital molecular ocupado más alto (HOMO) del otro reactivo. A veces esto solo es posible para el HOMO del estado π* fotoexcitado de un reactivo y generalmente la interacción es suprafacial (en la misma cara de los orbitales moleculares π. Sin embargo, tenga en cuenta que para las cicloadiciones de 2π + 2π, una reacción térmica es favorable si uno de los π-MOS interactúa antarafacialmente (en caras opuestas) y experimenta torsión de los dos extremos del MO en direcciones opuestas durante la cicloadición. Por razones estéricas, esto generalmente no es factible. Sin embargo, ocurre fácilmente para las cetenas (ver sección 3.4).
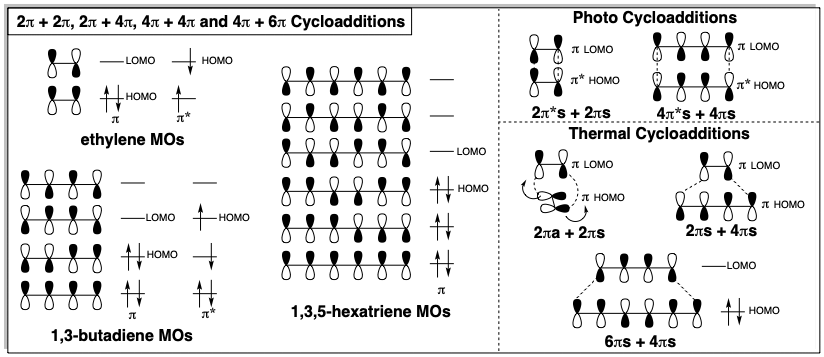
A continuación se presentan algunos ejemplos de luxaciones de retro cicloadición. Los precursores de 75 y 76 pueden cicloadirse con activación térmica mientras que los precursores de 77 y 78 pueden requerir fotoactivación, catálisis de metales de transición o implicar reacción antarafacial de un enlace p, por ejemplo, de una cetena.
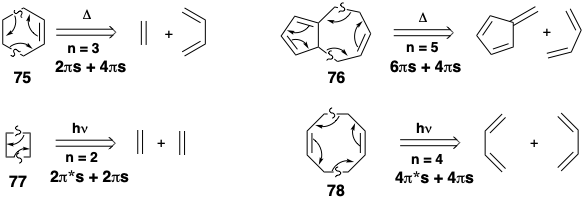
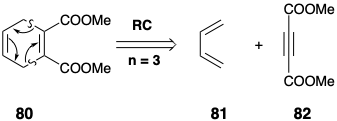
El circuito de 2n átomos puede contener más de n-2 enlaces π. Por ejemplo, la dislocación de 80 a 81 y 82 corresponde a una cicloadición que ocurrirá fácilmente térmicamente. El desplazamiento de dos enlaces σy n-2 = 1 enlace π escinde el anillo en dos polienos y n = 3 es un número impar.
Dado que el circuito 11,12,13,14,15 en PGE 1 es consonante, en teoría la anulación del anillo de ciclopentanona formando el enlace 11,12 se puede lograr explotando la activación polar proporcionada por los grupos funcionales en las posiciones 11 y 15. Por ejemplo, la conjugación con el carbonilo C-15 en 84 debería facilitar la generación de un carbanión en C-12 que podría acoplarse con el aldehído carbonilo en 84 para generar 83 directamente. Sin embargo, PGE 1 contiene una matriz sensible de β-hidroxiciclopentanona que se deshidrata fácilmente para dar PGA 1. Por lo tanto, las estrategias tempranas para la síntesis de PGE 1 estuvieron dominadas por los esfuerzos para enmascarar la matriz reactiva de β-hidroxicetonas.
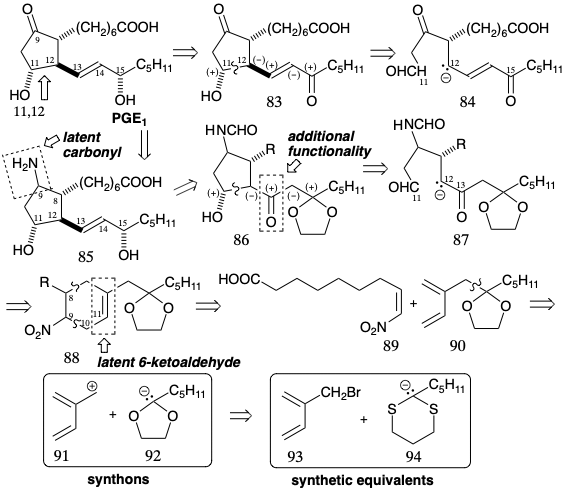
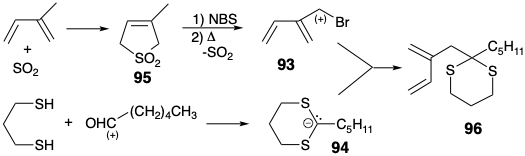
En la primera síntesis total de PGE 1, lograda por E. J. Corey 8, la cetona carbonilo C-9 se transportó en forma latente como un agrupamiento formamida relativamente no reactivo que se transformó en un grupo carbonilo solo al final de la síntesis, vide infra. Para la última conexión esquelética entre los carbonos 11 y 12 en un precursor 87 de 85 en la estrategia de Corey para PGE 1, la reactividad nucleofílica en C-12 es proporcionada por un grupo carbonilo agregado a C-13. Este carbonilo se eliminaría después de la ciclación polar a 86. Además, la reducción seguida de β-eliminación fomentada por un grupo carbonilo en C-15 generaría la insaturación requerida entre los carbonos 13 y 14. Los dos grupos carbonilo reactivos en 87 podrían derivarse de un precursor latente, el enlace C=C en un ciclohexeno 88. Una doble desconexión de este último intermedio sugiere el dieno 90 y el dienófilo 89 como precursores que proporcionarían 88 por una cicloadición de Diels-Alder. El uso de un grupo nitro en 88 y 89 como precursor para el grupo formamido en 87 viene dictado por la reactividad favorable de dienófilos deficientes en electrones hacia dienos ricos en electrones como 90. Para la síntesis del dieno 90, se eligió la unión polar de un sintón isoprenoide electrófilo 91 con un sintón carbanión acetal 92 ya que el bromuro de isoprenilo 93 está fácilmente disponible. Un carbanión 94 derivado de ditiano serviría como el equivalente sintético del sintón carbanión cetal numerado 92. El bromuro alílico 93 se preparó mediante bromación alílica por radicales libres de 95, un cicloaducto obtenido a partir de isopreno y dióxido de azufre. La abstracción de radicales libres del hidrógeno alílico se ve favorecida por la deslocalización del radical alílico resultante. La abstracción de hidrógeno bencílico y la bromación con N-bromosuccinimida (NBS) se ve favorecida de manera similar. Tanto el 95 como el bromuro alílico derivado son sólidos cristalinos a partir de los cuales se pueden generar los dienos correspondientes por cicloeliminación de\(\ce{SO2}\). La alilación de 94 con 93 entregó un dieno 96 que es un equivalente sintético de 90 (ver arriba).
En lugar de convertir primero el tiocetal 96 en cetal 90, el primero se hizo reaccionar con el dienófilo 97 para producir ciclohexeno 98. Después, el ditiocetal se reemplazó por un grupo protector de etileno cetal. La revelación de los grupos carbonilo latentes por escisión oxidativa del alqueno 88 proporcionó el ε-keto aldehído 87. La ciclación aldólica catalizada por base de 87 luego entregó 86 de manera estereoselectiva. Así, debido a que el sustituyente C-12 es epimerizable en 86, se produjo la relación trans requerida termodinámicamente favorecida entre las dos cadenas laterales voluminosas. Debido a que no se ejerció ningún control durante la generación del estereocentro C-11, también se formó parte del epímero equivocado.
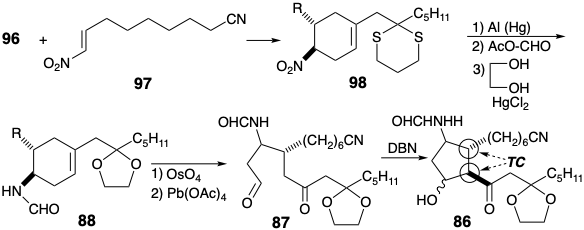
Aunque el esqueleto de carbono prostanoide se generó en la ciclación aldólica de 87 para dar 86, la finalización de la síntesis requirió un amplio ajuste de funcionalidad y nivel de insaturación. La matriz sensible de β-hidroxicetonas se acetiló inmediatamente y luego se redujo el grupo carbonilo para evitar la deshidratación. Después de la desprotección de la cetona y deshidratación del intermedio 99, el carbonilo alílico en una enona intermedia se redujo con\(\ce{Zn(BH4)2}\), un agente reductor de hidruro suave, para producir un alcohol alílico 100. La hidrólisis del nitrilo y el acetato fue seguida por la protección de los grupos hidroxilo en las posiciones 11 y 15 como éteres de tetrahidropiranilo. La hidrólisis posterior de la formamida requirió condiciones más vigorosas para entregar un derivado protegido con THP de 85.
La producción de PGE 1 por generación de la matriz sensible de β-hidroxicetonas en condiciones suaves se logró luego en el paso clave del plan sintético. La estrategia para usar un grupo amino como carbonilo latente en el precursor inmediato 85 de PGE 1 dependió de una secuencia de reacciones precedentes. Así, la oxidación selectiva a una imina se logró mediante N-bromación con NBS seguida de eliminación promovida por bases. Finalmente, la hidrólisis de los grupos protectores tanto de la imina como del éter de tetrahidropiranilo (THP) en condiciones ligeramente ácidas proporcionó PGE 1.
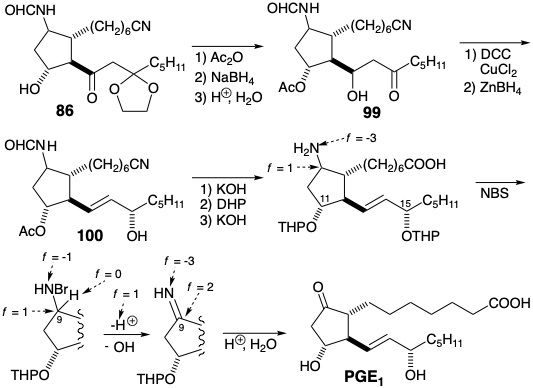
El proceso clave de oxidación se basa completamente en reacciones polares. Así, la N-bromación oxida el nitrógeno amino de f = -3 a f = -1. La eliminación de\(\ce{HBr}\) entonces reduce el nitrógeno a f = -3 mientras oxida carbono e hidrógeno.