3.4: Síntesis de Prostaglandinas a partir de Precursores Policíclicos
- Page ID
- 70186

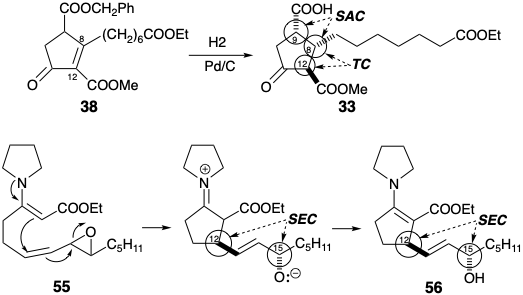
En las síntesis anteriores de prostaglandinas se logró el estereocontrol de varias maneras. Por ejemplo, la generación estereoselectiva de 33 de 38 dependió del control de aproximación estérica (SAC) durante la hidrogenación catalítica para favorecer una relación cis entre los sustituyentes en las posiciones 9 y 8. El posterior control termodinámico (TC) favoreció una relación trans entre los sustituyentes en las posiciones 9 y 12 mediante la epimerización de un intermedio cis termodinámicamente menos estable al isómero trans más estable. Se adujo el control estereoelectrónico (SEC) para dar cuenta de la generación estereoselectiva de la relación correcta entre los estereocentros en las posiciones 12 y 15 en 56 durante la ciclación de 55.
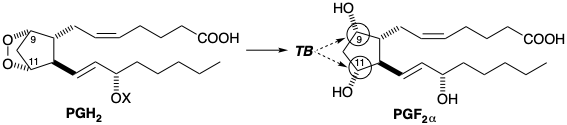
En esta sección se considerará otra técnica para lograr el estereocontrol. Así, la proximidad de los grupos funcionales puede estar asegurada por la táctica de unirlos en un anillo temporal que finalmente se escinde. La biosíntesis de PGF 2α implica tal puente temporal (TB) que impone una relación cis entre los átomos de oxígeno en las posiciones 9 y 11. Estos oxígenos están unidos con un enlace O-O en el intermedio prostaglandina endoperóxido PGH 2.
A menudo, los puentes temporales contienen grupos funcionales en forma latente. Anteriormente vimos que las cicloolefinas pueden ser disociadas oxidativamente para producir compuestos dicarbonilo. La creación del esqueleto PGF 2α por generación de ambos enlaces C=C a partir de precursores de carbonilo sugiere una subdiana de 1,5-dialdehído 101. Dado que el sustituyente aldehído en el estereocentro C-12 debe ser epimerizable, el aldehído cis 102 menos favorecido termodinámicamente también puede servir como subdiana. Los grupos carbonilo en 102 pueden ocultarse en forma latente en el puente insaturado temporal de 103. Mediante el empleo de un segundo puente insaturado temporal, se puede lograr una síntesis altamente estereocontrolada de PGF 2α. Así, la matriz cis-1,3-diol que se encuentra en PGF 2α y en el intermedio propuesto 103 se puede obtener —por oxidación Baeyer-Villiger de la metil cetona derivada- a partir del cis-diácido 104 que puede producirse por escisión oxidativa de endo - diciclopentadieno (DCPD). En esta estrategia para la síntesis de prostaglandinas 9, la generación de la relación cis requerida entre los estereocentros en las posiciones 8 y 9 depende en última instancia de una preferencia estereoelectrónica para la generación del isómero endo en lugar de exo de DCPD durante la dimerización\(2 π + 4 π\) cicloaditiva de 1,3-ciclopentadieno. Esto se ve favorecido por el solapamiento orbital secundario entre el enlace C=C “no participante” y el dieno cicloadista.
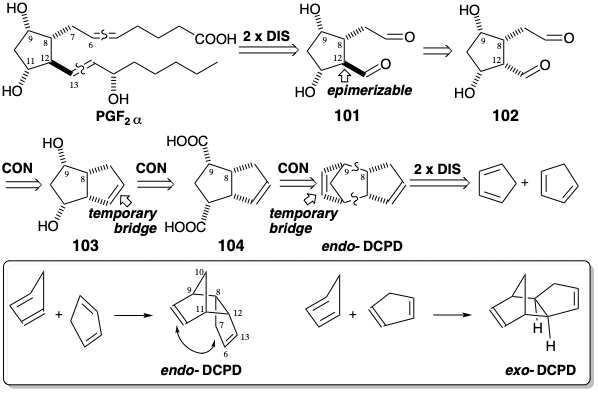
Esta síntesis explota la escisión selectiva de un puente temporal, el enlace C=C más tenso, para producir un dialdehído 105. Después de la conversión a dicetona 106, el enlace C=C restante se oxida parcialmente para suministrar 107 después de la acetilación. La oxidación de Baeyer-Villiger produce entonces un tetraacetato. La saponificación seguida de escisión oxidativa proporciona el dialdehído 102 de su precursor latente, la matriz de diol vecinal en 108. La epimerización de 102 en la posición 12 genera 101 que forma un hemiacetal en el que un aldehído carbonilo se enmascara adecuadamente para permitir la olefinación quimioselectiva del carbonilo restante para proporcionar 109. Para evitar la reducción del aldehído carbonilo, 109 se convierte en un acetal mixto antes de una reducción no estereoselectiva de la cetona carbonilo. La escisión reductiva del β-tricloroetil acetal en 110 luego permite la olefinación del grupo aldehído restante para proporcionar 111 y finalmente PGF 2α racémico junto con el 15-epímero racémico.
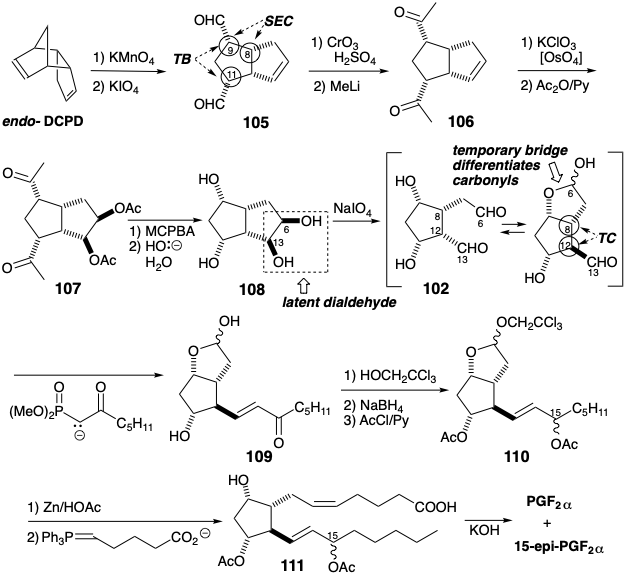
La segunda estrategia de Corey para la síntesis total de prostaglandinas 10 explota dos puentes temporales para asegurar las relaciones estereoquímicas adecuadas entre los estereocentros en las posiciones 9, 8 y 11. La relación cis entre los sustituyentes en las posiciones 8 y 9 en 101 está asegurada por un anillo temporal en el precursor de lactona 112 que se genera por funcionalización estereoselectiva de una olefina 113. Otro puente temporal, que invora el sustituyente C-8, se utiliza en la lactona 114 para asegurar una relación cis con el hidroxilo en la posición 11. Dado que las cetonas son ésteres latentes, la cetona cíclica 115 puede servir como precursora de 114. Se puede esperar la regioespecificidad requerida en la oxidación Bayer-Villiger de la cetona 115, ya que esta reacción implica la migración 1,2-a un terminal deficiente en electrones. El grupo que más fácilmente soporta una carga positiva parcial migra preferentemente. Así, en 115 el grupo alquilo secundario que también es alílico migra en preferencia al grupo alquilo primario. Una relación trans entre el sustituyente en la posición 12 y los estereocentros restantes en el anillo de ciclopentano es en última instancia la consecuencia del control de aproximación estérica durante la cicloadición de un precursor de 1,3-ciclopentadieno 5-sustituido 116 con cetena.
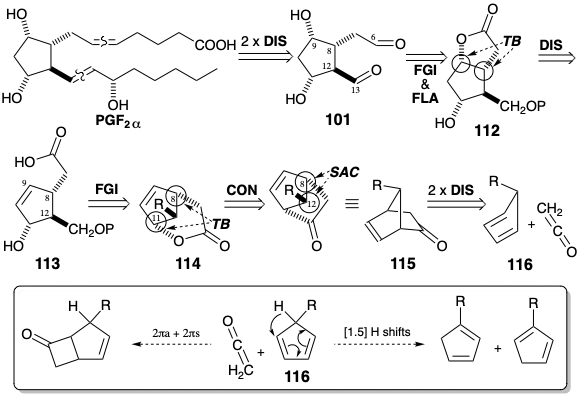
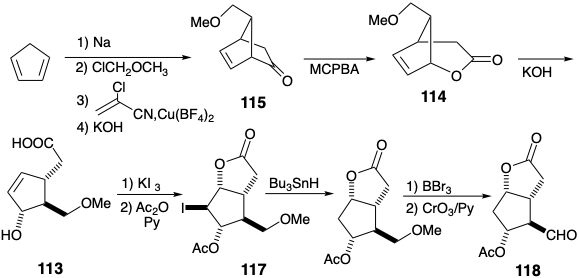
Una falla potencial en esta estrategia surge de la inestabilidad de 1,3-ciclopentadienos 5-sustituidos que se reordenan fácilmente a temperatura ambiente, por [1.5] migraciones sigmatrópicas de hidrógeno (ver sección 4.7), para generar mezclas de isómeros 1 y 2 sustituidos. Esta isomerización se eluyó utilizando\(\ce{Cu(BF4)2}\) para catalizar la reacción de Diels-Alder de 5-metoximetil-1,3-ciclopentadieno con α-cloroacrilonitrilo a baja temperatura (para evitar el reordenamiento del 1,3-ciclopentadieno 5-sustituido). Este cloronitrilo —una cetena latente— se somete a cicloadición de 2π +4π con ciclopentadieno mientras que la cetena prefiere someterse a cicloadición de 2πa + 2πs (ver sección 3.3). Otro defecto potencial, la epoxidación del enlace C=C en 115 durante la oxidación de Baeyer-Villiger, aparentemente se evita mediante el blindaje estérico del enlace C=C. La introducción estereoselectiva (una configuración se genera preferentemente en un nuevo estereocentro) la introducción del hidroxilo C-9 resulta de la influencia estereocontroladora de un puente temporal. Así, un nucleófilo que se une a C-8 en 113 se introduce intramolecularmente (nucleófilo interno) a un centro electrófilo creado en C-9 mediante la adición de I\({}^{\oplus}\) al enlace p C-C para dar 117. La desprotección y oxidación generan el aldehído 118.
El apéndice de la cadena lateral inferior al aldehído 118 se logra mediante la olefinación con un β-ceto-fosfonato. La reducción de una cetona intermedia seguida de transesterificación da un diol de lactona en el que el hidroxilo en la posición 9 se diferencia de los hidroxilos restantes. Estos últimos son luego enmascarados como éteres de tetrahidropiranilo (THP) en 119. La reducción parcial de la lactona y la olefinación de Wittig de un aldehído intermedio proporciona un intermedio clave 120 que puede convertirse en prostaglandinas E o F de las series “1" o “2" mediante manipulación apropiada de grupos protectores para permitir ajustes selectivos de funcionalidad y niveles de saturación. Así, el hidroxilo en la posición 9 en 120 o el derivado 122 puede oxidarse selectivamente para proporcionar PGE 2 o PGE 1 respectivamente mientras que la desprotección de 120 o 122 entrega PGF 2α o PGF 1α respectivamente.
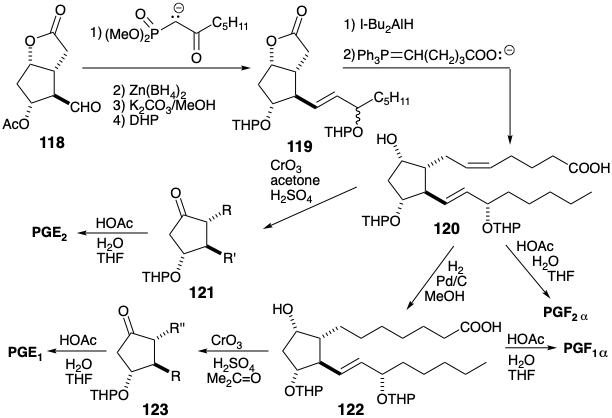
Una estrategia estrechamente relacionada para la síntesis de prostaglandinas 11 explota exactamente los mismos puentes temporales para imponer una relación cis entre los sustituyentes en las posiciones 9, 8 y 11 en el núcleo de ciclopentano. Sin embargo, un orden diferente para generar las mismas conexiones esqueléticas obvia la necesidad de usar grupos protectores. Así, la cadena lateral superior se agrega a un precursor 124. Sin embargo, la cadena lateral inferior ya está presente en un biciclo [2.2.1] hepteno intermedio 125 previo a la escisión de Baeyer-Villiger del puente temporal. También se evita la necesidad de una difícil cicloadición de Diels-Alder a baja temperatura mediante el uso de un fulveno 127 en lugar de un ciclopentadieno 5-sustituido para reaccionar con un equivalente de ceteno 128. La presencia de un aldehído enol acetato en 126 y 127 también evita la necesidad de un ajuste posterior del nivel de funcionalidad después de la eliminación hidrolítica del grupo enmascarante.
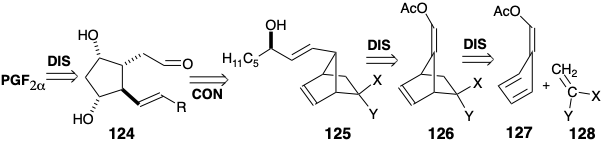
El acetato de enol en 129 es fácilmente hidrolizado selectivamente en presencia del α-cloronitrilo. La generación estereoselectiva de la configuración requerida en la posición incipiente 12 es, sin duda, consecuencia del control termodinámico. Así, el aldehído adopta la configuración menos congestionada estericamente. Después de la olefinación de este aldehído con un carbanión β-cetofosfonato, la cetona carbonilo se reduce mediante una reacción de Meerwein-Pondorf-Verly seguida de hidrólisis del α-cloronitrilo entregando 130 en buen rendimiento general. Dado que esta cetona incorpora una cepa de anillo apreciable, es posible una escisión inusual de tipo Bayer- Villiger con anión hidroperóxido. Los perácidos reaccionan con 130 para dar epóxidos, pero el anión hidroperoxi reacciona exclusivamente con el grupo carbonilo. El diol 131 se puede enmascarar para eventualmente permitir la oxidación selectiva de los PGE de suministro de 9-hidroxilo mientras que la elaboración adicional a PGF 2α sigue de cerca la síntesis de 120, excepto que los grupos protectores de THP son innecesarios. Debido a que no se requieren pasos que impliquen la introducción o eliminación de grupos protectores, esta estrategia sintética es notablemente eficiente.

Todas las estrategias presentadas hasta el momento tuvieron éxito. Desafortunadamente, los intentos fallidos a menudo no se publican. En ocasiones se describen en tesis doctorales. Sería un error suponer que la planeación sintética para la síntesis total de moléculas complejas es tan confiable, incluso para las superestrellas indiscutibles de la síntesis orgánica. Por lo tanto, para mantener una perspectiva realista, consideraremos algunas estrategias defectuosas de vez en cuando.
La estrategia defectuosa de R. B. Woodward
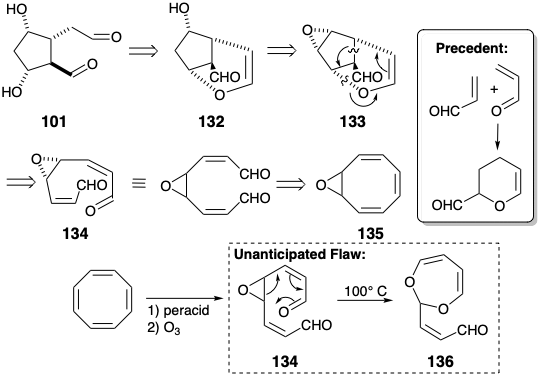
El dialdehído 101, que incorpora toda la información estereoquímica requerida para el anillo ciclopentano de las prostaglandinas, podría derivarse de un enol éter 132 en el que dos grupos funcionales están enmascarados internamente en un puente temporal. Se recomienda el uso de un epóxido 133 como precursor del alcohol 132 por la consiguiente posibilidad de que 133 pueda generarse a partir de un precursor simétrico 134. Esta estrategia requiere el descubrimiento de un método para lograr la escisión reductora regioselectiva del epóxido 133 para generar 132. La presencia en 133 de un anillo de seis miembros que contiene un enlace C=C sugiere la posibilidad de una cicloadición 2π + 4π que generaría 133 a partir de 134. La cicloadición hetero intramolecular de Diels Alder de un aldehído dienófilo a un aldehído α, β-insaturado dieno para generar el anillo de dihidropirano en 133 está precedida por la correspondiente dimerización intermolecular de acroleína que, sin embargo, favorece la orientación incorrecta. La generación de 134 podría ser factible mediante la escisión oxidativa selectiva del enlace C=C más rico en electrones en el monóxido de ciclooctatetraeno 135. Esta estrategia, ideada por Woodward, es fatalmente defectuosa porque el intermedio 134 se somete a un nuevo reordenamiento homo retro Claisen produciendo 136 en lugar de la deseada cicloadición de Diels-Alder. 1
Estrategias de contracción de anillos
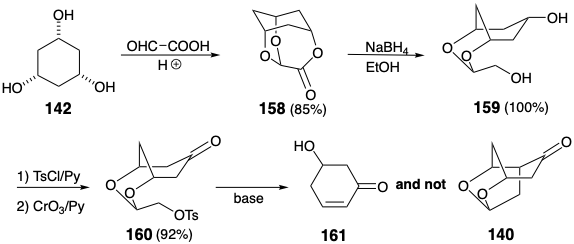
El acetal mixto 137 es otro precursor temporalmente puenteado similar al aldehído 118 de Corey (ver arriba) excepto que el nivel de funcionalidad en 137 es idéntico al de 101. Por lo tanto, el puente temporal en 137, como el de 132, es un derivado enmascarado internamente de dos grupos funcionales a diferencia de un precursor latente. Woodward 12 ,13 reconoció que un precursor potencial de 137 es 138 en el que C-11 y el carbono aldehído carbonilo están temporalmente puenteados por un enlace C-C. Los intermedios 137 y 138 son isómeros con diferentes conectividades y una distribución diferente de funcionalidad pero con un nivel de funcionalidad global idéntico. El reordenamiento de 138 a 137 implica la oxidación a un aldehído de un carbono que porta un hidroxilo y la reducción concomitante de un carbono que porta un grupo saliente electronegativo. Tal proceso, un reordenamiento de pinacol, es impulsado a su finalización por la generación energéticamente favorable de un doble enlace C=O a expensas de dos enlaces simples C-O. La subdiana 138 puede simplificarse por dislocación a un precursor 139 menos funcionalmente sustituido que podría proporcionar 138 por adición 1,2-dioxidativa. La generación del enlace C=C en 139 a partir de una cetona en 140 se sugiere con el objetivo de desconectar y reorganizar funcionalmente esta subdiana a un precursor simétrico 141 por dislocación polar que implica la desconexión de un electrófilo interno. La cetona carbonilo en 140 puede proporcionar reactividad nucleofílica para facilitar la formación de este enlace C-C. Un puente temporal en 141 entre el electrófilo y el nucleófilo asegura la necesaria relación cis entre los hidroxilos enmascarados en 141 y el enlace C-C recién creado en 140. En última instancia, 141 podría razonablemente estar disponible mediante la protección selectiva de dos de los tres grupos hidroxilo idénticos en todos los cis 1,3,5- ciclohexanotriol y la oxidación del hidroxilo restante.
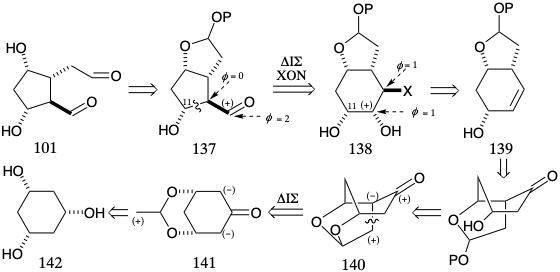
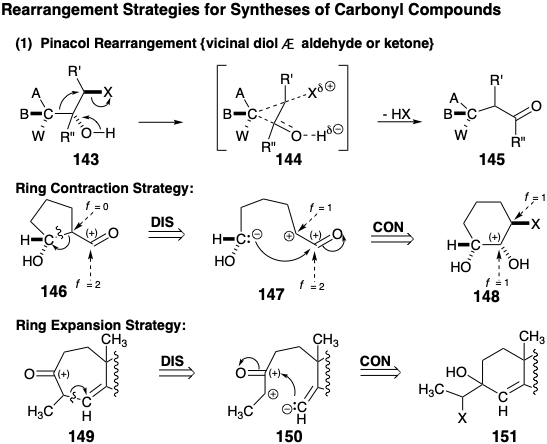
Estereoespecífica (la configuración estereoquímica del isómero del producto está determinada por la del isómero reactivo) se espera la generación de las relaciones estereoquímicas requeridas en 137 durante el reordenamiento del pinacol de 138. Por lo tanto, tales reordenamientos, es decir 143 a 145, generalmente proceden con la retención de la configuración en el carbono migratorio debido a un estado de transición temporalmente puenteado 144.
Análisis reterosintético en dos pasos de reordenamientos polares
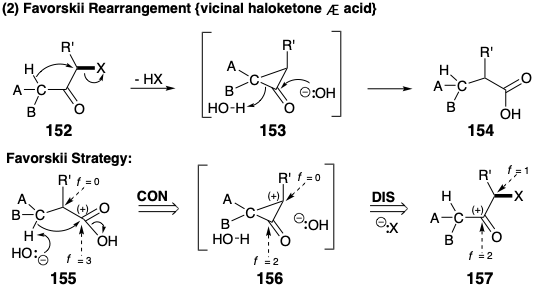
La dislocación de un producto 146 de reordenamiento de pinacol a un precursor 148 puede verse como una desconexión polar del carbono migratorio como nucleófilo dando como resultado la oxidación del extremo de migración. La posterior conexión del carbono migratorio nucleofílico da como resultado una reducción del origen de la migración (nótese que este es un nucleófilo interno). De hecho, los pasos de desconexión y conexión ocurren simultáneamente en los reordenamientos del pinacol. Sintéticamente el reordenamiento 148 a 146 da como resultado la contracción del anillo. Los reordenamientos del pinacol también pueden resultar en la expansión del anillo. Esto se ejemplifica retrosintéticamente mediante la generación de un precursor 151 con un anillo de seis miembros para una diana 149 con un anillo de siete miembros por desconexión a 150 y posterior conexión a 151. Este ejemplo, que también sugiere que un carbono vinílico puede servir como grupo migratorio, es un paso en una estrategia para la síntesis total del terpeno longifoleno que será considerada en el capítulo 4. Por supuesto, los reordenamientos de pinacol también pueden ocurrir en sistemas acíclicos. El reordenamiento Favorskii de α-halocetonas para generar ácidos carboxílicos acíclicos o de anillo contraído está estructural y funcionalmente relacionado con el reordenamiento del pinacol. Sin embargo, el reordenamiento de Favorskii implica un intermedio temporalmente puenteado en lugar de un estado de transición. Así, 1,3-eliminación de 152 genera un intermedio de ciclopropanona 153 a partir del cual se forma un producto 154 de anillo contraído por escisión inducida por nucleófilos. Retrosintéticamente, los reordenamientos Favorskii generan un precursor 156 más conectado a partir de una diana de ácido carboxílico 155. La desconexión del precursor 156 sugiere entonces el precursor 157 reorganizado estructural y funcionalmente en el que el nivel de funcionalidad del grupo carboxilo (f = 3) es igual a la suma de los niveles de funcionalidad de una cetona carbonilo (f = 2) y un carbono portando un grupo de salida electronegativo (f = 1). Así, al igual que en el reordenamiento del pinacol, el reordenamiento de Favorski no da como resultado ningún cambio neto en el nivel de funcionalidad molecular, es decir, ninguna oxidación o reducción neta. Más bien, estos procesos implican la redistribución de la funcionalidad mediante un proceso redox intramolecular. Por lo tanto, las dislocaciones de reordenamiento son complejas porque implican etapas de conexión y desconexión acopladas, así como una redistribución asociada de la funcionalidad.
Aunque la diferenciación de los tres hidroxilos en 142 se logró por reacción con ácido glioxálico, la segunda estrategia de Woodward también fue fatalmente defectuosa (ver sección 3.5). La escisión reductora de 158 proporcionó el diol 159 cuantitativamente, y el hidroxilo primario en 159 se activó fácilmente de manera selectiva por tosilación. Sin embargo, la cetona 160, obtenida por oxidación del monotosilato, no logró producir 140 tras el tratamiento con una variedad de bases. En cambio, la eliminación de un grupo β-alcoxi para producir 161 ocurrió con la exclusión completa de la alquilación intramolecular.
Para obviar la necesidad de generación de enolato β a los grupos alcoxi como en 141, 162 fue reconocido como precursor directo de la olefina 139. La desconexión polar de 162 sugiere la adición de un electrófilo de carbono a un doble enlace C=C como en 163. Las dos dislocaciones, 140 a 141 y 162 a 163 son isoelectrónicas (mecanismos con patrones de movimiento de electrones idénticos). Es decir, ambos implican el movimiento de dos pares de electrones y la escisión de un enlace σ-C-C. Sin embargo, mientras que la escisión de un enlace C-O en un equivalente sintético de 141 para dar 161 es impulsada por la producción de un enlace C=O, la escisión similar de un enlace C-O en un equivalente sintético de 163 generaría un intermedio relativamente inestable, un alílico carbocatión.
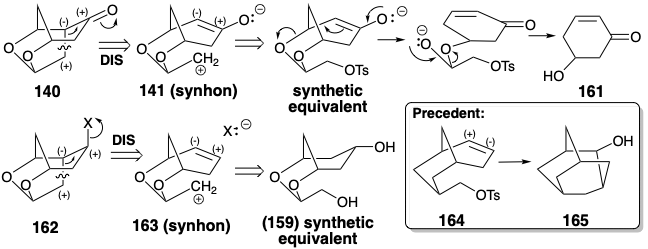
Los equivalentes sintéticos del sintón asimétrico 163 podrían obtenerse del diol simétrico 159 disponible por β-eliminación. Desafortunadamente, un precedente sugería que la ciclación de 163 podría no ocurrir de la manera deseada. Así, el análogo carbocíclico 164 produce el sistema de anillo isomérico 165 tras la solvolisis. Sin embargo, se podría argumentar que los sustituyentes alílicos de oxígeno en 163 podrían desfavorecer tal modo de ciclación que debe generar una deficiencia de electrones β a los sustituyentes alcoxi.
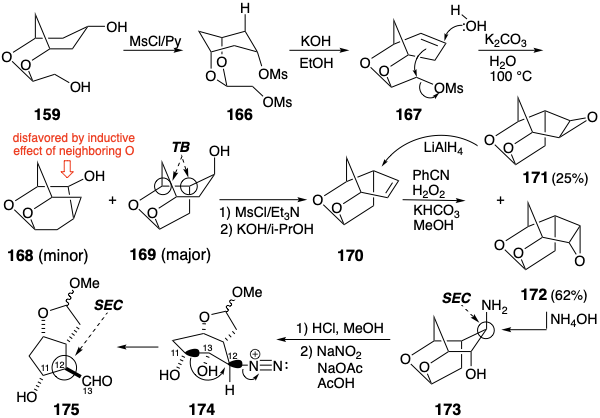
Esta vez la apuesta dio sus frutos. 13 El bismesilato 166 de 159 proporcionó la olefina 167 tras la eliminación selectiva del grupo mesiloxi secundario. La solvolisis de 167 produjo una mezcla que contenía solo 5-8% del producto no deseado 168. Por supuesto, el producto de ciclación deseado 169 es racémico. Se puede resolver. Sin embargo, solo un enantiómero conduce a prostaglandinas de configuración natural. Así, si bien esta síntesis (vide infra) resuelve “el principal problema esteroquímico inherente a la síntesis de prostaglandina F 2α — el alineamiento de los cuatro átomos quirales contiguos en el resto ciclopentano”, el proceso no es enantioselectivo. La mitad del intermedio 169 es el enantiómero equivocado, que no es fácilmente reciclable. La deshidratación del producto 169 de ciclación apropiado proporcionó luego 170. La preferencia estereoquímica requerida durante la adición 1,2-dioxidativa a 170 se logró mejor con el ácido perimídico generado in situ a partir de benzonitrilo y peróxido de hidrógeno. Esta reacción suministró una mezcla del exo epóxido 171 y el endoepóxido 172. El epóxido anterior podría reciclarse a 170. Apertura nucleofílica controlada estereoelectrónicamente del anillo epoxi en 172 suministró 173 estereoespecíficamente. Este isómero es obligatorio para el posterior reordenamiento de la contracción del anillo similar al pinacol que se produce tras la desaminación del acetal mixto bicíclico 174. Así, la migración del carbono C-11 incipiente se ve favorecida en 174 por una relación antiperiplanar (dos enlaces o grupos que se encuentran en un mismo plano con un ángulo diedro de 180°) con el grupo saliente. El reordenamiento de 174 ocurre no solo con la retención de la configuración en el carbono migratorio, sino que también genera estereoespecíficamente la configuración requerida en C-12 en el producto 175 debido a la inversión de Walden durante la reacción intramolecular de S N 2. La posterior elaboración del intermedio clave 175 en prostaglandinas siguió reacciones bien precedentes.
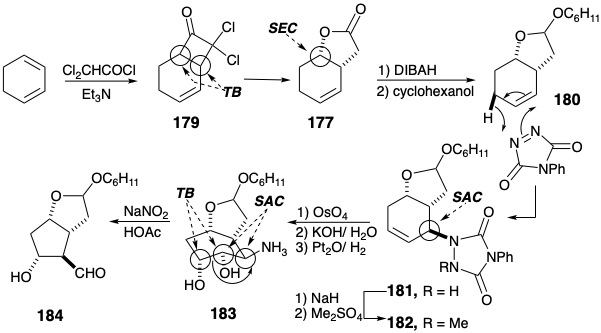
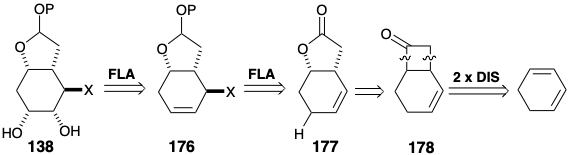
Corey ideó una ruta alternativa 14 a intermedios clave del tipo 138. Así, 138 podría estar disponible por adición cis -1,2-dioxidativa al precursor de alqueno 176. Además, se podría anticipar una relación trans entre el grupo saliente incipiente X y los átomos de oxígeno recién introducidos sobre la base del control de aproximación estérica. El control del enfoque estérico también podría favorecer la relación trans requerida entre X y el anillo de lactona. Esto sería especialmente cierto si la oxidación alílica se lograra mediante un mecanismo ene que implicara un estado de transición cíclica estéricamente exigente. Además, dicho mecanismo aseguraría el regiocontrol requerido, es decir, la sustitución con reordenamiento alílico, durante la introducción de X. La lactona en 177 podría obtenerse a partir de un precursor latente, la cetona en 178, disponible a su vez por una cicloadición de 2π + 2π de cetena a un precursor simétrico, 1,3-ciclohexadieno.
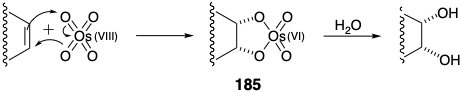
La cicloadición estructuralmente selectiva (ver sección 3.3) de diclorocetona a 1,3-ciclohexadieno liberó 179 de los cuales se obtuvo la lactona insaturada 177 por decloración reductora seguida de oxidación de Baeyer-Villiger (ver sección 3.5). La reducción parcial seguida de cetalización suministró 180 que de manera estereoselectiva proporcionó 181 tras la reacción de eno con N-feniltriazolindiona. La hidroxilación del derivado 182 también procedió estereoselectivamente. Esta última reacción también involucra un anillo temporal, un éster de osmato 185, que impone una relación cis entre los átomos de oxígeno recién introducidos.