
3.5: Síntesis de Prostaglandinas a partir de Ciclopentanos
- Page ID
- 70185

Dado que la estereoquímica de todos los sustituyentes en el anillo debe preferirse termodinámicamente para derivados de ciclopentano como PGE 1, un método de estereocontrol menos potente que el uso de puentes temporales parecería adecuado para la síntesis de prostaglandinas. Además, la disponibilidad de precursores simples de ciclopentanoides incluyendo ciclopentadieno, que se utilizó en muchas de las síntesis descritas anteriormente, condujo a la formulación de una estrategia simple para la síntesis total estereocontrolada de prostaglandinas. Además, tales estrategias son adecuadas para la síntesis total enantioselectiva (ver sección 3.6). El análisis de reactividad polar de PGE 1 como en 186 sugiere la dislocación de esta diana estereoquímicamente compleja en dos fragmentos 187 y 188 que contienen solo un estereocentro cada uno. 15 Así, el control del enfoque estérico podría favorecer la ddición del nucleófilo vinil 188 desde la cara menos congestionada del anillo de ciclopentanona, la cara opuesta a un sustituyente en la posición 11.
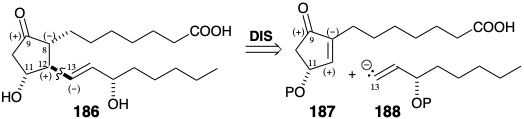
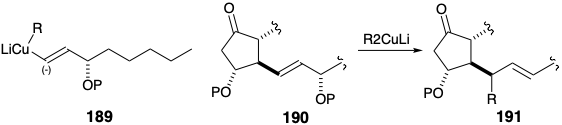
Además, las 1,4-adiciones de diorganocupratos de litio como 189 con cetonas α, β-insaturadas son especialmente susceptibles a dicho SAC. Sin embargo, también se sabía que los diorganocupratos de litio desplazaban el oxígeno alílico como el de la posición 11 en la ciclopentenona 187 o en la posición 15 en la cadena lateral inferior de la prostaglandina como en la hipotética reacción de 190 para generar 191, un subproducto inútil. ¿Debería tal defecto potencialmente fatal evitar una mayor consideración de una estrategia sintética? La respuesta depende del valor del posible descubrimiento de que la falla no es fatal. Esto lleva a que se agregue otra regla general a la lista iniciada en la sección 1.5:
(6) Favorecer estrategias potencialmente defectuosas solo si el esfuerzo que implica un mayor examen de la posible falla se compensa con la recompensa potencialmente grande de una síntesis especialmente elegante y eficiente.
La estrategia de Woodward que implica la cicloadición intramolecular de 134 para generar 133 es un ejemplo de que este principio no da sus frutos. La estrategia de Woodward que implica la alquilación intramolecular de la cetona 160 y su éxito final en lograr la conexión esquelética requerida mediante una modificación de la estrategia es un ejemplo de otro principio de planeación sintética:
(7) Diseñar estrategias de respaldo, especialmente para pasos riesgosos.
El circuito 9,8,12,11-en 187 es disonante. Una estrategia para la generación de esta matriz funcional disonante (ver página 88) implica la adición 1,4-dioxidativa (cicloadición 4πs + 2πs) de oxígeno singlete a un precursor de 1,3-ciclopentadieno monosustituido 194 para generar un endoperóxido 192 que podría sufrir desproporcionación a la requirió hidroxiciclopentenona en analogía con la desproporción de PGH a PGE. 15 La alquilación del anión ciclopentadienuro con bromoéster 195 produciría un 1,3-ciclopentadieno 5-sustituido. Sin embargo, el isómero 2-sustituido requerido está fácilmente disponible porque los monoalquil 1,3-ciclopentadienos existen a temperatura ambiente como una mezcla de equilibrio de principalmente isómeros 1 y 2 sustituidos que se forman a partir del isómero 5-sustituido por [1.5] migraciones sigmatrópicas de hidrógeno.
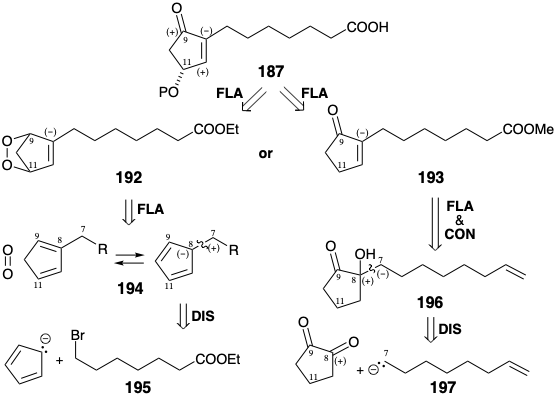
Alternativamente, una simple ciclopentenona monosustituida 193 podría convertirse en 187 por oxidación alílica. Una ruta al 193 se sugiere por la posibilidad de que el enlace C=C en esta enona se pueda producir a partir de una ciclopentanona 196 por eliminación de agua. Si el grupo saliente es un hidroxilo, la presencia de dicha funcionalidad en la posición 8 en un precursor 196 invita a una dislocación adicional a un sintón nucleofílico de la cadena lateral superior 197 y un electrófilo de carbonilo, 1,2-ciclopentandiona. La funcionalidad carboxilo electrófila en 193 está latente en 197 para evitar la reacción intramolecular no deseada con el centro nucleófilo en la posición 7.
Es interesante señalar que las dos rutas a 187 señaladas anteriormente implican estrategias polares electrónicamente complementarias para generar el enlace 7-8. Una vía explota un electrófilo de cadena lateral superior y un nucleófilo de ciclopentilo (es decir, 195 y anión ciclopentadienuro) mientras que la otra vía explota un nucleófilo de cadena lateral superior y un electrófilo de ciclopentilo (es decir, 197 y 1,2-ciclopentandiona).
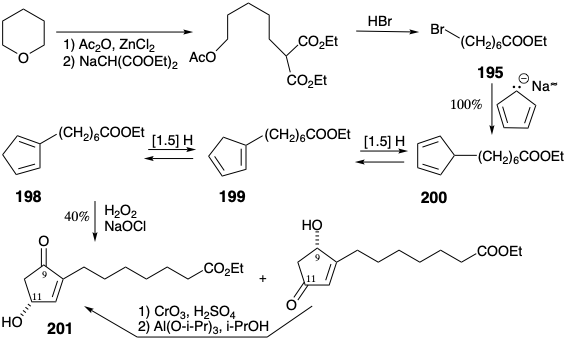
El bromoéster 195 se preparó a partir de tetrahidropirano y malonato de dietilo. El oxígeno singlete, generado químicamente, reacciona con los ciclopentadienos monosustituidos 198-200 en condiciones básicas para suministrar hidroxiciclopentenona 201 y su isómero que tiene un hidroxilo en la posición 9 y un carbonilo en la posición 11. Este último isómero se convirtió fácilmente en 201 mediante una secuencia de oxidación y reducción.
Se preparó un reactivo de Grignard equivalente sintético de sintón nucleofílico 197 mediante monohidroboración de 1,7-octadieno seguido de yodo-deborinación y reacción del yoduro resultante 202 con magnesio. La oxidación de ciclopentanona proporciona 1,2-ciclopentandiona cuyo metil enol éter 203 suministró ciclopentenona 204 tras la reacción con yoduro de 7-octenil-magnesio seguido de hidrólisis del enol éter y deshidratación. La generación de un éster a partir del precursor latente requirió la escisión oxidativa selectiva de un doble enlace C=C en 204. Esto se logró fácilmente mediante la epoxidación del enlace C=C más rico en electrones con perácido seguido de la escisión oxidativa de 205 con peryodato. La metilación suministró el éster 190 que se bromó alílicamente para proporcionar 207 después de la hidrólisis de un bromuro intermedio 206.
Un nucleófilo vinílico de cadena lateral inferior se prepara por hidroaluminación de (S) -1-octin-3-ol (208) seguido de yododealuminación de un vinil alano intermedio para entregar yoduro de vinilo 209 ópticamente puro de configuración absoluta correcta. Este yoduro también está disponible por cloroacilación de acetileno con cloruro de valerilo seguido de yododecloración de un cloruro de vinilo intermedio para suministrar yodocetona 211 que proporciona 209 racémico tras la reducción de borohidruro. La resolución del 209 racémico se puede lograr con la sal fenetilamina del derivado hemiftalato. El grupo hidroxilo en 209 debe ser enmascarado antes del intercambio de litio-yodo. La reacción de 209 con etil vinil éter proporciona un derivado α-etoxietilo (EE) 212 que proporciona un cuprato divinil 213 por intercambio metal-halógeno con t-butillitio seguido de la adición de\(\ce{CuI}\) y\(\ce{Bu3P}\).
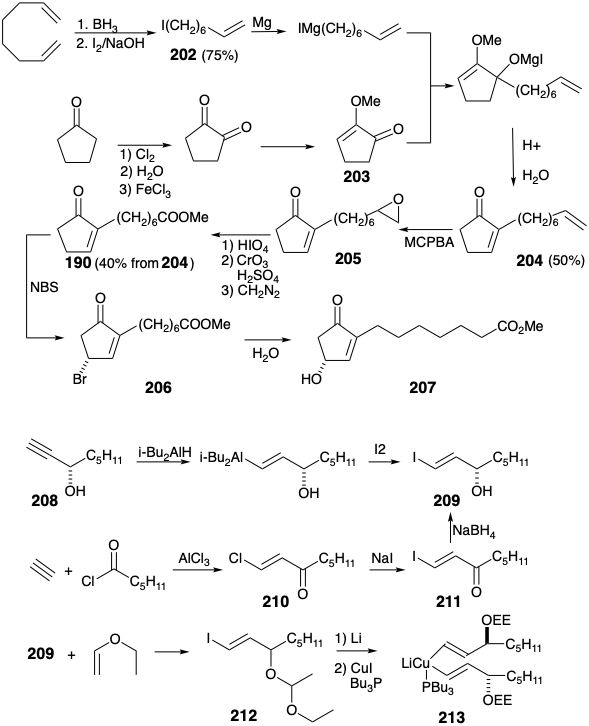
La adición clave de 1,4-cuprato de divinilcuprato ópticamente puro 213 al derivado THP 214 de la hidroxiciclopentenona racémica 201, seguida de la eliminación de los grupos protectores THP y EE, proporciona una mezcla casi 1:1 de éster etílico 215 (-) -PGE 1 con el estereoquímica absoluta del producto natural y su diastereómero que es epimérico en las posiciones 8, 12 y 11. La hidrólisis del éster para producir PGE 1 se pudo lograr en condiciones especialmente suaves por incubación con levadura de panadería. La reacción del cuprato 214 de divinil ópticamente puro con 214 ópticamente puro (ver sección 3.6) entrega (-) -PGE 1 exclusivamente. 16

Otra estrategia para la síntesis de prostaglandinas a partir de precursores de ciclopentano 17 explota el control de abordaje estérico durante la reducción con hidruro de un derivado 216 de PGE 2 para proporcionar la configuración correcta en la posición 9 en PGF 2α. El análisis polar de 216 sugiere que la cadena lateral superior puede ser anexada por reacción de un nucleófilo cis vinil 218 con una cetona α, β-insaturada 217. El análisis polar de 217 sugiere una dislocación adicional al enolato de cetona 219 y formaldehído. Se podría lograr una síntesis regioselectiva del enolato requerido mediante escisión reductora de α-bromo cetona 220. La funcionalización apropiada de la olefina 221 podría ser factible a través de la adición 1,2-dioxidativa. Que 221 pueda obtenerse estereoselectivamente a través de la apertura nucleofílica regioselectiva de monóxido de ciclopentadieno (223) por un nucleófilo vinil 222 es la consecuencia razonable de un mecanismo S N 2 con ataque al enlace alílico C-O más débil. Así, la escisión de un puente temporal, el epóxido, procederá con la inversión de la configuración en un extremo que conduce a una relación trans entre los grupos nucleófilo y nucleófugo que se convierten en los sustituyentes en las posiciones 12 y 11 respectivamente.
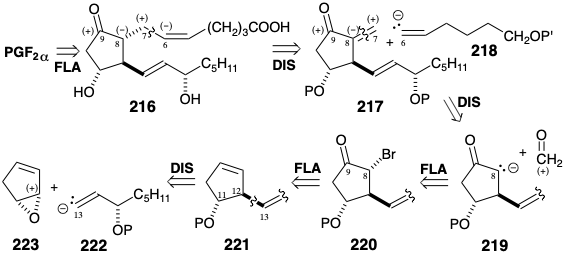
El acetiluro de litio de 3- (α-etoxietoxi) -1-octin puede servir como equivalente de carbanión vinílico terminal. Así, reacciona con el epóxido 223 para proporcionar 224 después de la bencilación de un alcóxido intermedio. La hidrólisis del grupo protector EE seguida de reducción trans estereoselectiva con hidruro de un alcohol propargílico intermedio en presencia de metóxido seguido de enmascaramiento del alcohol alílico resultante proporciona 225. Esa bromación hidroxi de 225 ocurre estéreo y regioselectivamente aparentemente resulta de una preferencia estérica por el ion α-bromonio 226 que es atacado por el agua en la posición menos estérica congestionada, es decir, alejada del sustituyente voluminoso en la posición 12, entregando 227. Que el enlace ciclopenteno C=C reaccione con preferencia al enlace C=C de la cadena lateral es una consecuencia del efecto desactivador de extracción de electrones del sustituyente alílico de oxígeno. La oxidación de 227 a la cetona correspondiente seguida de una reacción de Perkow suministra el derivado enol 228 regioespecíficamente. La generación de un enolato a partir del 228 por reacción con t-butil-litio activa regioespecíficamente la posición 8 para la reacción nucleofílica con formaldehído que suministra 229. Esta condensación aldólica es promovida por un puente temporal que es proporcionado por un catión quelante de zinc. La deshidratación de 229 proporciona entonces la enona 230 que agrega un cuprato de vinilo cis 231 para producir la cadena lateral superior en 232 después de la hidrólisis selectiva del grupo protector EE y la oxidación del alcohol primario. La reducción de hidruro estereoselectiva, es decir SAC, de 232 proporciona PGF2α después de la eliminación reductora de los grupos protectores de éter bencílico y benciloximetílico en 233.
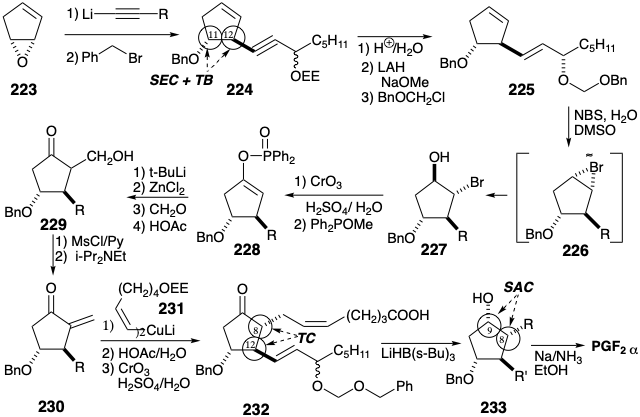
La apertura estereoespecífica de epóxidos por nucleófilos de carbono puede explotarse para introducir ambas cadenas laterales prostanoides en un núcleo de ciclopentano. Una estrategia notable para la síntesis total de PGF 2α a partir de ciclopentadieno 18 primero simplifica la diana mediante la desconexión de la cadena lateral superior de la manera habitual en el enlace C=C. El paso clave en la estrategia implica el desplazamiento regioselectivo S N 2 de un electrófilo en la posición 12 por un sintón de carbanión transvinílico de cadena lateral inferior nucleófilo 222. La relación trans requerida entre los sustituyentes en las posiciones 11 y 12 está asegurada por un puente epóxido temporal en 235 entre los estereocentros en las posiciones 11 y 12. Este epóxido podría generarse a partir del correspondiente monotosilato de trans diol 236. La introducción de un fragmento nucleofílico de la cadena lateral superior también podría lograrse estereoespecíficamente mediante un ataque S N 2 sobre un epóxido 237, un electrófilo simétrico que contiene un puente temporal entre las posiciones incipientes 8 y 12. La generación estereoselectiva de 237 podría lograrse mediante el control de aproximación estérica durante la epoxidación de un precursor ciclopenteno 238. Finalmente, la relación cis entre los sustituyentes de oxígeno en 238 se puede asegurar mediante un tercer puente temporal, esta vez entre dos átomos de oxígeno en un precursor de endoperóxido 239 que está disponible a partir de 1,3-ciclopentadieno por cicloadición de 2π + 4π de oxígeno singlete.
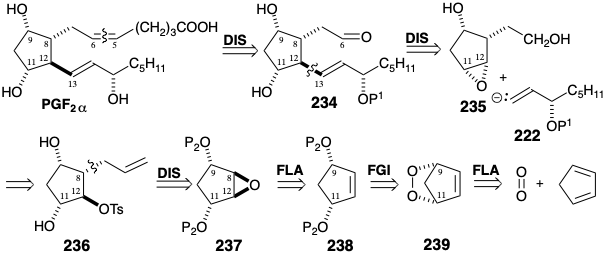
La escisión reductora del puente de peróxido temporal en 239 entrega un cis diol. El regiocontrol durante la escisión del epóxido intermedio 242, que podría lograrse con un acetiluro de aluminio, aparentemente resulta de un puente temporal entre nucleófilo y electrófilo. Así, el sustituyente hidroxi-etilo en 242 reacciona con el nucleófilo organoalano. El alcoxialano entonces suministra el nucleófilo alquinilo intramolecularmente como en 243 a la posición deseada 12 y no a la posición 11. El hidroxilo primario en el tetraol 238. Finalmente, la relación cis entre los sustituyentes de oxígeno en 238 se puede asegurar mediante un tercer puente temporal, esta vez entre dos átomos de oxígeno en un precursor de endoperóxido 239 que está disponible a partir de 1,3-ciclopentadieno por cicloadición de 2π + 4π de oxígeno singlete.

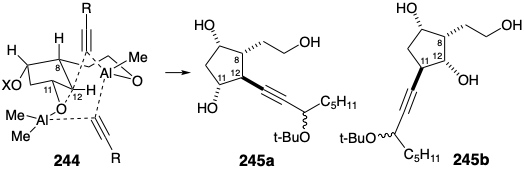
Se postuló que el regiocontrol durante el ataque nucleofílico al epóxido intermedio 235 podría ser proporcionado por un puente temporal entre el nucleófilo y el electrófilo. Así, el sustituyente hidroxietilo en 235 reaccionaría con un nucleófilo de organoalano. El alcoxialano resultante puede suministrar el nucleófilo alquinilo intramolecularmente como en 244 a la posición deseada 12 y no a la posición 11. De hecho, esta reacción dio el aducto regioisomérico deseado 245a con un rendimiento del 60% y ninguna traza del regioisómero indeseado 245b. Un apoyo adicional para esta explicación mecanicista es proporcionado por la observación de que la sililación de 235 antes de la reacción con el alano produjo una mezcla regioisomérica de aductos e incluso favoreció el ataque nucleofílico en la posición 11 por 2. 6:1. También es digno de mención el hecho de que el puente que involucra al grupo hidroxietilo y al nucleófilo acetiluro en 244 se fusiona de manera trans con el anillo ciclopentano, mientras que el puente epóxido está cis fusionado. Así, mientras que los anillos pequeños prefieren las fusiones cis, las fusiones trans pueden no ser deformadas e incluso favorecidas termodinámicamente para anillos más grandes.
Para completar el esqueleto de prostaglandina, el hidroxilo primario en el tetraol intermedio 242 se diferenció por tritilación. Después de la acetilación de los hidroxilos restantes y la detritilación, el alcohol primario resultante 243 se oxidó a un aldehído antes de la adición final de la porción restante de la cadena lateral superior de la manera habitual.
En la estrategia anterior para la síntesis de prostaglandinas, la activación polar que es potencialmente proporcionada por la funcionalidad relacionada con la diana no se explota para la construcción esquelética. Más bien, la electrofilicidad en las posiciones 8 y 12 es proporcionada por la funcionalidad añadida, los epóxidos en 235 y 240. La economía de la funcionalidad se sacrifica a favor de incorporar grupos de salida temporalmente puenteados que aseguren el estereocontrol.