
3.6: Síntesis Enantioselectiva de Prostaglandinas
- Page ID
- 70174

Todas las síntesis totales anteriores de prostaglandinas produjeron estos productos naturales junto con sus isómeros enantioméricos no naturales porque cada síntesis comenzó con materiales de partida no asimétricos o racémicos y emplearon reactivos racémicos o no asimétricos. En algunos casos, las mezclas racémicas de precursores enantioméricos se separaron por resolución y luego se convirtieron en el producto natural enantioméricamente puro. Pero este enfoque de la síntesis total de productos naturales no racémicos quirales suele ser inherentemente derrochador ya que la mitad del precursor racémico, el enantiómero equivocado, debe descartarse. Muy raramente, el enantiómero equivocado puede convertirse en el enantiómero correcto o convertirse en el producto natural en una síntesis enantio-convergente mediante una secuencia de reacción única.
Existen tres tácticas que permiten la síntesis enantioselectiva de productos naturales. Todos ellos dependen de la quiralidad de los productos naturales para proporcionar materiales de partida asimétricos o para inducir asimetría durante la generación de intermedios quirales a partir de precursores proquirales. En la discusión posterior se presentarán ejemplos de síntesis que son enantiocontroladas por el uso de: (1) reactivos quirales no racémicos; (2) metabolismo microbiano, un caso especial de categoría 1; o (3) materiales de partida quirales no racémicos.
Enantiocontrol mediante un reactivo quiral no racémico
En la síntesis discutida anteriormente, la quiralidad de 235 está determinada por un intermedio 246 que se generó por reacción de un nucleófilo alílico con el epóxido proquiral 240. Dado que el reactivo alilante empleado era no asimétrico, el producto quiral fue racémico. El intermedio 235 también se ha preparado de otra manera, una que genera solo el enantiómero correcto requerido para la síntesis total de prostaglandinas naturales ópticamente puras. 19 En esta síntesis asimétrica, la quiralidad de 235 está determinada por un intermedio 247 producido por hidroboración del dieno proquiral 248 con un dialquil borano quiral no racémico.
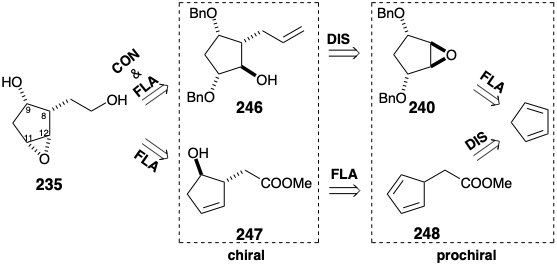
El 1,3-ciclopentadieno 248 5-sustituido se generó a -78°C por alquilación de ciclopentadienuro de sodio y se trató in situ con (+) -di-3-pinanilborano, seguido de peróxido de hidrógeno alcalino para dar hidroxiéster 247, que fue al menos 96% ópticamente puro. Debido al control de aproximación estérica durante la adición sin de hidruro de boro a 248 y la posterior retención estereoespecífica de la configuración durante el reemplazo oxidativo de boro con oxígeno, el nuevo estereocentro en la posición 9 (numeración PG) en 247 se genera estereoselectivamente con el configuración antinatural, opuesta a la requerida para las prostaglandinas. Sin embargo, la configuración cis requerida se genera fácilmente por la inversión de S N 2 del hidroxilo, y la lactonización proporciona 249. La generación estereoselectiva del epóxido cis en 235 depende de la influencia de un puente temporal. Así, el hidroxilo en C-9 dirige la entrega de oxígeno por enlaces de hidrógeno con MCPBA como se muestra en 250. Este puente temporal en el estado de transición de una reacción es un ejemplo de un efecto de grupo vecino.
Enantiocontrol por Metabolismo Microbio
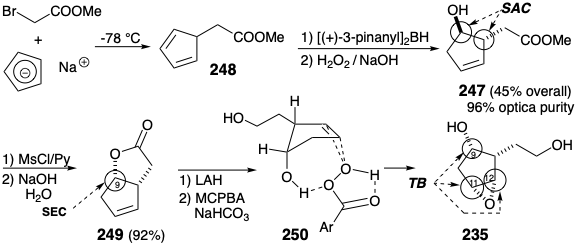
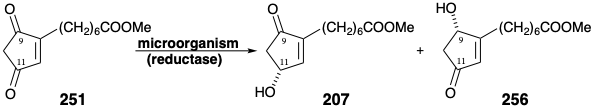
Las enzimas que catalizan las oxidaciones y reducciones microbianas son moléculas quirales no racémicas que a menudo promueven transformaciones altamente enantioselectivas de sustratos proquirales sintéticos. 20 Se han explorado varias estrategias para la generación microbiana enantioselectiva de intermedios de hidroxiciclopentenona para síntesis de prostaglandinas. Por ejemplo, el intermedio quiral 207 podría estar disponible por oxidación alílica microbiana del precursor proquiral 190 o por reducción microbiana del precursor proquiral 251. La diona 251 debe estar fácilmente disponible por oxidación de la mezcla de hidroxiciclopentenona obtenida por desproporción catalizada por base de un cicloaducto 252 de oxígeno singlete 2π + 4π con ciclopentadieno 253.
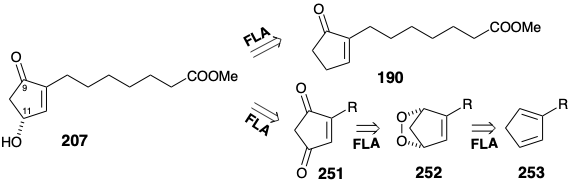
Una falla en la estrategia de oxidación alílica resultó de una proclividad general de los hongos que contienen hidroxilasas a degradar la cadena lateral carboxílica de 190 por el mecanismo de escisión β-oxidación-retro Claisen discutido en la sección 3.1. Así, la escisión de una unidad de acetato produjo 254 mientras que 255 se generó tras la pérdida de un segundo acetato.
Se espera una proclividad de la diona 251 hacia la monorreducción debido al efecto activador de la retirada opuesta de electrones por dos grupos carbonilo conjugados. Por lo tanto, se prevé que la monorreducción catalizada por enzimas de 251 sea fácilmente alcanzada por microorganismos. Sin embargo, dos problemas interfirieron con los intentos de obtener una síntesis bioorgánica asimétrica práctica de 207 enantioméricamente puro a partir de la α-dicetona 251 vinilógica. Primero, la reducción del enlace C=C a menudo acompañó a la reducción de C=O. La saturación de cetonas α, β-insaturadas es una transformación microbiológica común. Esta reacción secundaria no deseada se evitó mediante la reducción microbiológica de 251 en presencia de exceso de 2-ciclohexenona o metil vinil cetona como sustratos competitivos para las reductasas de enlace C=C pero no para las reductasas C=O. 16 Además, con una amplia variedad de microorganismos, la reducción del grupo incipiente 9-ceto que genera 256 compitió con la reducción deseada para generar 207 con un hidroxilo en la posición 11 incipiente.
Una estrategia alternativa menos directa para la síntesis enantioselectiva de 207 mediante reducción de carbonilo asimétrica explota la funcionalidad y el ajuste del nivel de insaturación de un precursor de hidroxidiona 257 que podría estar disponible por enantioselectivo (generando un enantiomérico puro producto de un precursor aquiral) reducción de una triona 258. El análisis polar de 258 sugiere una síntesis de esta matriz funcional disonante mediante la condensación de una metil cetona 259 con el diéster disonante, dietil oxalato.
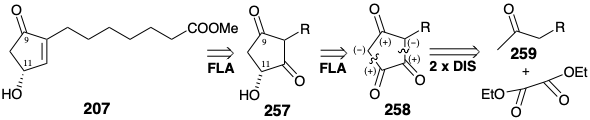
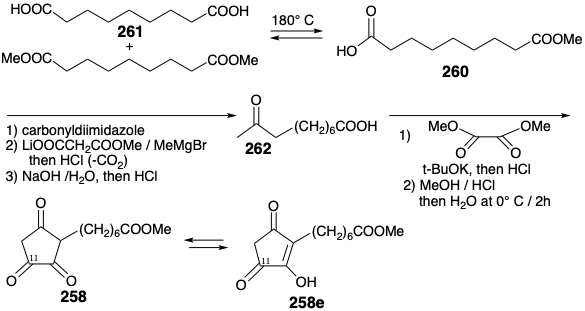
La triona 258 realmente existe como un enol 258e que es un derivado hidroxi de 251. Pero se puede esperar que este enol tenga menos proclividad que 251 hacia la saturación porque el sustituyente hidroxilo reducirá la electrofilicidad del enlace C=C mediante la donación de un par de electrones no enlazantes. Además, el grupo carbonilo en la posición 11 incipiente en 258e debería ser especialmente susceptible al ataque nucleofílico por hidruro debido al efecto dipolo de un grupo hidroxilo vecinal. Se inició una síntesis de triona 258 con ácido azelaico (260) y su éster dimetílico para proporcionar azelato de monometilo (261) por equilibrio térmico. La condensación de la imidazolida derivada con el enolato de magnesio de monometil malonato de litio seguida de descarboxilación, hidrólisis y una segunda descarboxilación proporcionó metil cetona 262. La condensación de Claisen con oxalato de dimetilo y la posterior ciclación de Dieckmann y metilación del grupo carboxilo produjeron el intermedio clave 258.
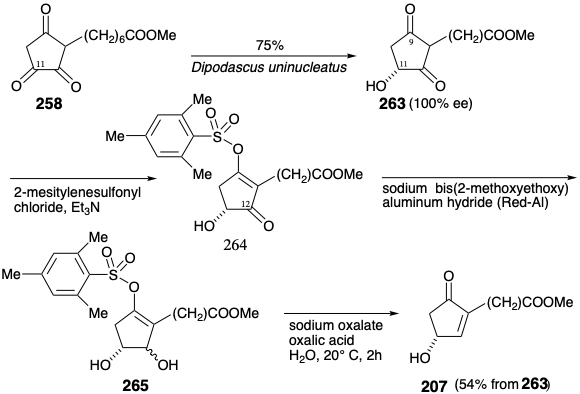
La triona 258 se redujo limpia y regioselectivamente a hidroxidiona 263 por una variedad de microorganismos. Dipodascus uninucleatus catlayó la reducción completamente asimétrica de 258 al alcohol 11 (R) que se requiere para la síntesis total de prostaglandinas. En contraste, Mucor rammanianus redujo 258 al alcohol 11 (S). La conversión de hidroxidiona 263 ópticamente pura en la hidroxiciclopentenona 207 ópticamente pura requirió la reducción selectiva del carbonilo en la posición 12. Esto se logró enmascarando selectivamente el carbonilo en la posición 9 como un enol mesitilenosulfonato 264 seguido de reducción con hidruro (ver sección 3.7). Posterior reordenamiento alílico hidrolítico del alcohol alílico intermedio 265 entregó 207. 16
Enantiocontrol mediante la explotación de materiales de partida no racémicos quirales
En lugar de usar la asimetría de productos naturales, por ejemplo enzimas, para inducir asimetría durante la conversión de precursores proquirales en intermedios sintéticos quirales, la asimetría de productos naturales fácilmente disponibles, por ejemplo azúcares, se puede incorporar en dianas sintéticas mediante conversión en no racémicos quirales. intermedios para síntesis totales. Para 207, el centro quiral en la posición 11 podría derivarse de un centro quiral en un azúcar. Dado que cada carbono en un azúcar está oxigenado, la desconexión polar de 207 quiral nonracémico con la condición límite de descubrir un segmento quiral derivado del azúcar sugiere un precursor trihidroxi 266 (ver sección 3.7). Nótese que 266 es un sintón nucleofílico umpolado generado por inversión de reactividad polar (PRI) en el incipiente carbonilo de la posición 9. Una mayor desconexión polar sugiere un nucleófilo 267 estabilizado con α-carbometoxi y D-gliceraldehído como electrófilo. El D-gliceraldehído debe estar disponible por escisión oxidativa de cualquier D-azúcar. Una síntesis especialmente eficiente es sugerida por una dislocación que implica acoplamiento reductor para conectar dos moléculas de gliceraldehído. El precursor axialmente simétrico D-manitol está disponible por reducción de D-manosa.
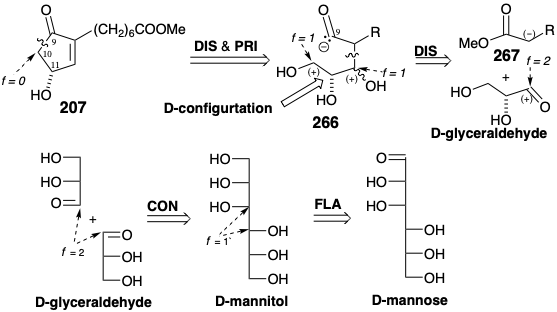
En una síntesis enantioespecífica de 207 ópticamente puro a partir de D-manitol, el oleato de metilo proporcionó el nucleófilo correspondiente a 267. 21 El enlace C=C en el oleato de metilo comprende un carboxilo latente que no se desenmascara hasta el final de la síntesis. La condensación aldólica de oleato de metilo con acetónido 269 de D-gliceraldehído entrega 270. La descetalización desenmascara una matriz de dihidroxilo vecinal (ver sección 3.7) y la posterior lactonización diferencia los hidroxilos primarios y secundarios explotando un grupo enmascarante interno y la estabilidad favorable de una butirolactona temporalmente puenteada. El hidroxilo primario libre en 271 se activa por tosilación, y el éter α-etoxietílico (EE) de una cianohidrina se genera a partir de la lactona carbonilo por reducción, formación de cianohidrina y O-alquilación con etil vinil éter. La ciclación de 272 luego completa el esqueleto de carbono. La eliminación de los grupos protectores, la escisión oxidativa del enlace C=C de la cadena lateral y la metilación del ácido carboxílico resultante, la hidrólisis de la cianohidrina y la deshidratación, proporciona entonces la hidroxi-ciclopentenona 207 ópticamente pura de 273.
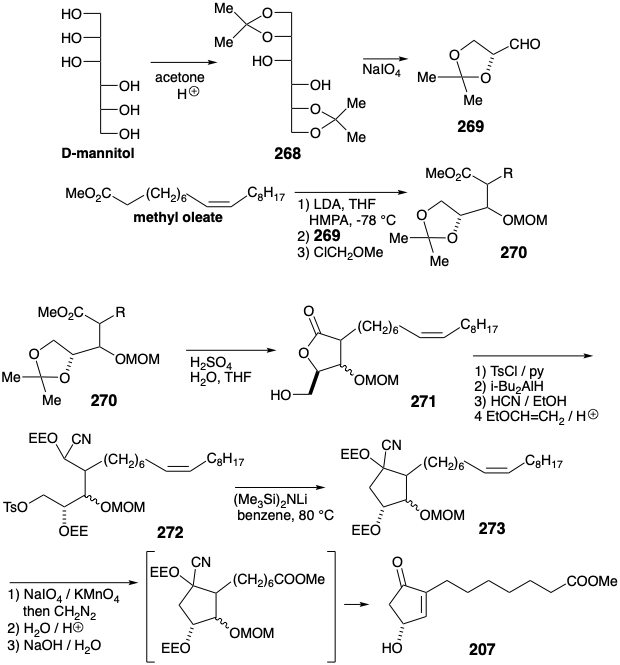
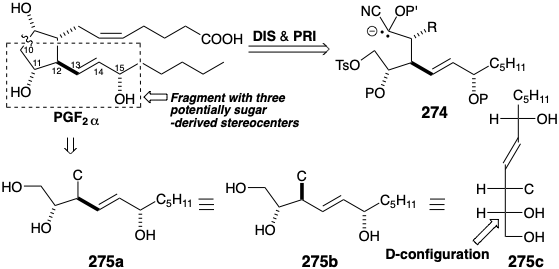
La abundante información funcional (oxígeno en cada carbono) y estereoquímica presente en los azúcares sugiere una síntesis aún más ambiciosa de prostaglandinas: incorporación de varios centros de quiralidad relacionados con el azúcar a partir de un material de partida de azúcar con un hidroxilo relacionado con azúcar en la incipiente posición 10 de PGF 2α. Así, la desconexión polar de PGF 2α con la condición límite de descubrir un segmento homoquiral derivado de azúcar sugiere un precursor 274 en el que la reactividad polar relacionada con la diana implicada por el hidroxilo en la posición 9 en PGF 2α debe invertirse (PRI), e.g., como carbanión estabilizado con nitrilo derivado de una aldehído cianhidrina. Las representaciones generalizadas 275 de 274, especialmente la proyección Fischer 275c enfatizan similitudes estructurales con precursores de D-azúcar.
La necesidad de desprotonar un éter de cianohidrina para generar 274 sugiere la sustitución del carboxilo terminal por un grupo funcional equivalente latente menos reactivo como un éter en 276 que también incorpora un éster como precursor de la cianhidrina en la incipiente posición 9 con vistas a una mayor desconexión polar de 276 a 278 se sugiere mediante análisis polar que también revela la posibilidad de desconectar 278 a un nucleófilo de acetato de metilo, o un carbanión de éster malónico en el que el grupo éster añadido sirve como control de reactividad elemento (CEA), y un electrófilo derivado del azúcar 279. La estereoquímica requerida en la posición 12 incipiente en 278 se generaría por inversión estereoespecífica durante la sustitución nucleofílica de una electrofuga de oxígeno por un carbanión. Esta estrategia puede ser descarrilada por una alternativa posible sustitución nucleofílica S N 2' del nucleófugo alílico en 2 7 9. Una dislocación alternativa de la subdiana 278 evita esta ambigüedad. El enlace sigma entre los carbonos incipientes 8 y 12 en 278 podría reemplazar un enlace sigma entre el éster incipiente carbonilo oxígeno y el carbono 14 por un proceso que implica el reordenamiento alílico de dos enlaces π y un enlace σ-en un precursor 280 por un desplazamiento cíclico de tres pares de electrones. Dichas reorganizaciones de bonos, conocidas generalmente como reordenamientos sigmatropicos [3.3], implican un puente temporal en el estado de transición que, para el 280 a 278, podría esperarse que la conversión adopte una conformación tipo silla (SEC) como en 281. En consecuencia, el reordenamiento implica una transferencia predecible de quiralidad desde el origen de migración en la posición 14 en 280 hasta el extremo de migración en la posición 12 en 278. La fuerza impulsora para los reordenamientos sigmatropicos es un aumento neto en la estabilidad termodinámica.
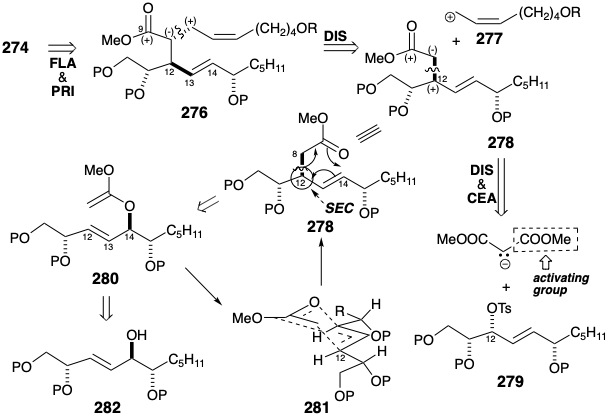
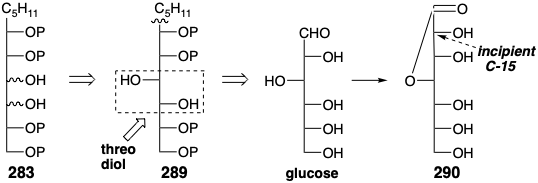
Para la conversión 280 a 278, un reordenamiento enol Ester-Claisen, esta ventaja energética se deriva de la generación de un bono C=O a expensas de un bono C=C. El acetal ceteno 280 es un derivado del alcohol alílico 282. Un progenitor similar al azúcar 283 para 282 se sugiere mediante la adición 1,2-dioxidativa. Dicho intermedio podría producirse, por ejemplo, por adición nucleofílica de un nucleófilo n-pentil a D-glucosa.

La conversión del diol 283 en trans alqueno 282 debe superar varios obstáculos. Dado que el diol vecinal está rodeado por grupos hidroxilo o derivados de grupos hidroxilo como en 284, la escisión reductiva que genera un estado intermedio o de transición que se asemeja al carbanión 285 podría conducir a la β-eliminación del oxianión vecinal incorrecto produciendo 287 más bien que 286.
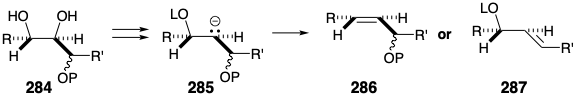
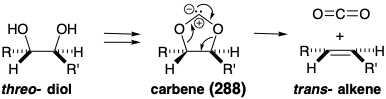
Un resultado más cierto se puede asegurar empleando una reacción alternativa, una cicloeliminación concertada de dióxido de carbono de un derivado 288 con puente de carbeno, para generar el trans alqueno requerido a partir de un diol vecinal. Tal proceso implica el desplazamiento cíclico de tres pares de electrones —dos enlaces σ-y un par de electrones no enlazantes en el carbono de carbeno— y es impulsado por la creación de dos enlaces C=O. Dado que la cicloeliminación es concertada, el derivado de carbeno generado a partir de un treo diol necesariamente se fragmenta a un alqueno trans mientras que el derivado de un eritro diol se fragmentaría para dar un alqueno cis. Por lo tanto, para ser precursor de un alqueno trans, el intermedio 283 debe incorporar un treo diol como en 289 y glucosa.
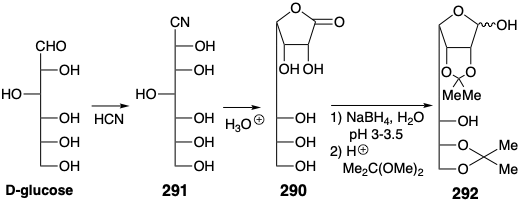
Sin embargo, la generación de 289 no necesita necesariamente proceder directamente a partir de la glucosa mediante la adición de un anión n-pentil. De hecho, la D-glicero-D-guloheptosa (290), que se puede preparar a partir de glucosa e incorpora la configuración requerida en la posición incipiente 15, está disponible comercialmente.
Se realizó una síntesis enantioespecífica de PGF quiral no racémico 2α que deriva tres centros de quiralidad de la glucosa. 22 Así, la glucosa se extiende en cadena por un carbono por adición de\(\ce{HCN}\). La hidrólisis catalizada por ácido y lactonización del intermedio cianohidrina 291 produce D-glicero-D-guloheptono-1,4-lactona (290). Los grupos hidroxilo en este intermedio deben diferenciarse para permitir la manipulación selectiva. Cuatro grupos hidroxilo se pueden enmascarar por cetalización con acetona. La posterior reducción parcial del grupo lactona proporciona un lactol 292 que proporciona 293 tras una reducción adicional y acetilación selectiva de un hidroxilo primario en presencia de dos hidroxilos secundarios. La matriz de treo diol que permanece desenmascarada en 293 ahora puede eliminarse estereoespecíficamente para generar un alqueno trans.
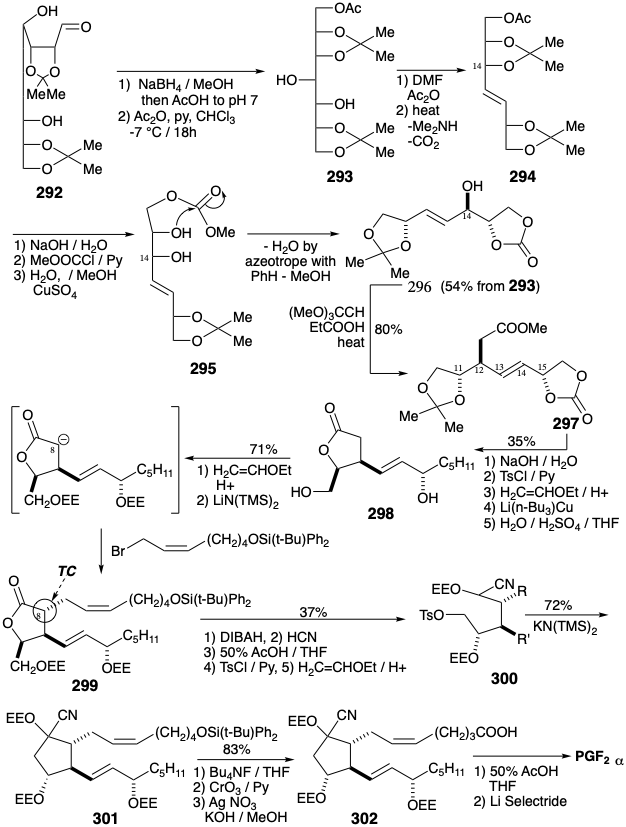
El intermedio de carbeno puenteado requerido se genera por termólisis de un acetal cíclico de dimetilformamida derivado del 293 en presencia de anhídrido acético. Una cicloeliminación concertada de este carbeno luego entrega estereoespecíficamente el trans-alqueno 294 requerido en aproximadamente 40% de rendimiento global de 290. El hidroxilo alílico enmascarado en la incipiente posición 14 ahora debe desenmascararse selectivamente para establecer el escenario para un reordenamiento orto Ester-Claisen. Pero tanto este hidroxilo como su vecino vecino están enmascarados en 294 por el mismo acetónido. Para capturar selectivamente a su vecino después de la eliminación del acetónido, el acetato en 294 se convirtió inicialmente en un carbonato de metilo que luego acila intramolecularmente el hidroxilo vecino en 295. Uno de los dos hidroxilos está así protegido por el puente temporal de un carbonato. El hidroxilo restante en 296 se desplaza luego con reordenamiento alílico por un grupo carbometoximetilo. Así, una reacción de Claisen ortoéster de 296 transfiere estereoespecíficamente la quiralidad de la posición 14 sustituida con hidroxilo en 296 a una posición 12 sustituida con carbono en 297. Este intermedio incorpora tres de los cinco centros quirales así como el enlace 13,14-trans C=C del PGF 2α diana. El apéndice de la cadena lateral carboxílica, después de enmascarar los hidroxilos como éteres α-etoxietílicos (EE), se logró mediante la alilación de un enolato de éster entregando 299. La estereoquímica en la posición 8 incipiente en 299 es una consecuencia del control termodinámico (TC) que favorece una relación trans entre los sustituyentes vecinales en el anillo de lactona de cinco eslabones. La anulación del anillo de ciclopentano requirió reducción parcial de la lactona carbonilo, formación de cianohidrina, eliminación de los grupos protectores EE, monotosilación selectiva del hidroxilo primario y protección del triol resultante como éteres EE. Finalmente el tratamiento de 300 con base generó el correspondiente carbanión estabilizado con nitrilo el cual se sometió a alquilación intramolecular proporcionando 301. Luego se generó un carboxilo después de la eliminación del grupo protector sililo. La eliminación de los grupos protectores EE del ácido carboxílico 302 proporcionó una cianhidrina que se escindió a la cetona correspondiente y se redujo estereoselectivamente in situ para producir PGF 2α. Esta eficiente intercepción de la cetona carbonilo es una táctica notable. El carbonilo se redujo para evitar la pérdida del hidroxilo en la posición 11 por deshidratación del intermedio PGE 2 sensible a bases.