4.4: Síntesis de Longifolene
- Page ID
- 70291

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Análisis Polar de Precursores Funcionalizados
El análisis de reactividad polar no es muy útil para la dislocación de longifoleno porque no tiene funcionalidad polar. Sin embargo, se sugieren precursores funcionalizados considerando posibles síntesis del metileno exocíclico. Así, un alcohol podría producir 32 por deshidratación, y el alcohol podría surgir por adición de un nucleófilo metílico a la cetona 43.
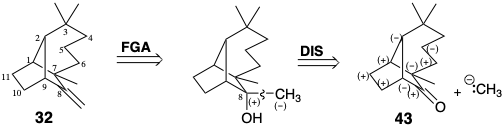
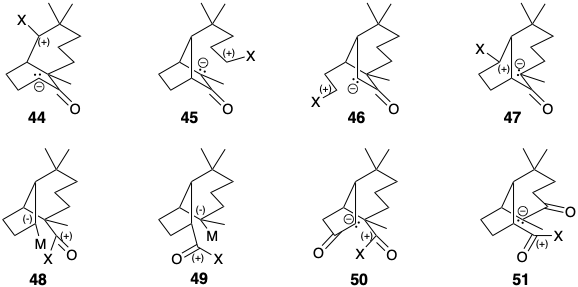
Para una síntesis conectiva C-C polar directa de 43, cualquier funcionalidad activadora polar adicional en un precursor debe perderse durante la formación del enlace C-C. Así, 44 - 47, precursores para una síntesis directa de 43, resultan de las cuatro posibles desconexiones que explotan la nucleofilia potencial del carbono a a un carbonilo, mientras que 48 y 49 resultan de las dos posibles desconexiones que explotan la electrofilicidad de un carbono carbonilo. Sin embargo, puede ser ventajoso utilizar una estrategia indirecta, una que incorpore funcionalidad adicional en los penúltimos intermedios de la construcción esquelética (eq. 50 ó 51).
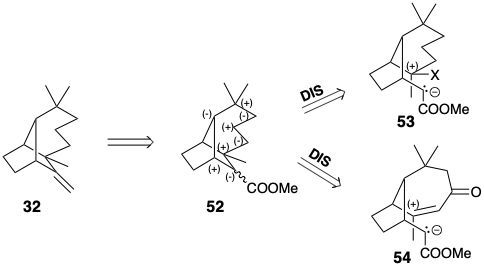
Esa funcionalidad debe entonces ser eliminada después de la finalización de la red de carbono. Por ejemplo, debido a que el metileno exocíclico de 32 podría derivarse razonablemente de un éster 52, otro conjunto de intermedios que tiene un patrón de reactividad diferente al primer conjunto puede generarse mediante análisis de reactividad polar (por ejemplo, 53 y 54). Se pueden generar muchos precursores adicionales considerando dislocaciones que involucran grupos activadores adicionales o insaturación.
Análisis topológico
Para moléculas como 32, que tienen una funcionalidad mínima y esqueletos complejos, se ha sugerido otro enfoque para identificar dislocaciones útiles. Así, la atención se dirige primero a “un análisis exhaustivo de las propiedades topológicas de la red de carbono para definir la gama de posibles precursores... a partir de los cuales se puede producir el esqueleto deseado mediante el establecimiento de uno o dos enlaces de conexión”. 3 Posibles reacciones, funcionalidad activadora apropiada, etc., sólo se consideran después del análisis topológico.
En muchos casos, las dislocaciones más útiles sintéticamente son el resultado de eliminar un enlace entre los átomos miembros del anillo, llamados átomos comunes, que están unidos a otros tres o cuatro miembros del anillo (pero no a dos). Para el longifoleno (32), en el que los átomos comunes están numerados 1-4, esto genera tres estructuras topológicamente simplificadas 55 - 57.
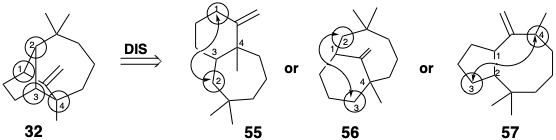
Otra serie útil se genera al eliminar un enlace entre un átomo común y un átomo no común. Dos miembros de esta serie son 58 y 59. Dado que algunas reacciones generan dos nuevos enlaces, por ejemplo, la cicloadición de Diels-Alder, se deben considerar las estructuras generadas al eliminar dos enlaces de la red original 32, especialmente que unen dos átomos adyacentes a uno o más átomos comunes como en 60. Sin embargo, los intermedios sugeridos por dislocaciones que implican la eliminación de un enlace entre átomos no comunes no pueden ignorarse a priori.
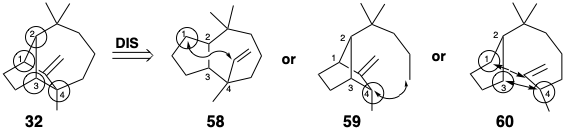
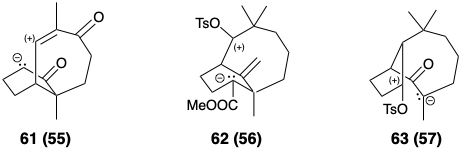
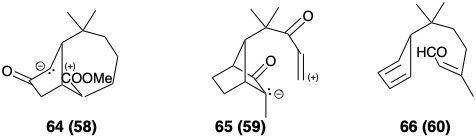
Después del análisis topológico, se consideran reacciones específicas y funcionalidad apropiada para permitir la formación de enlaces. El proceso se repite hasta que se genera una serie de precursores potenciales para cada penúltimo intermedio y así sucesivamente hasta que se completa el árbol sintético. A medida que se agrega funcionalidad a los intermedios, el análisis topológico se vuelve menos relevante. La “utilización máxima de la funcionalidad relacionada con (sub) objetivos” (ver sección 1.2) y, por lo tanto, el análisis de reactividad polar (ver sección 1.4) , se convierte en un factor importante en la planificación sintética. Los compuestos 61 - 66 son posibles derivados funcionalizados correspondientes a las estructuras 55 - 60, respectivamente.
En algún momento, se hace una elección entre un amplio abanico de posibilidades. Debe necesariamente ser “en gran medida una función de la metodología de química sintética disponible en su momento, de ciertas consideraciones prácticas como la disponibilidad de los materiales y reactivos necesarios, y de ciertos juicios subjetivos relacionados con la viabilidad de reacciones clave o la existencia de alternativas.” 3
Estrategias fatalmente defectuosas
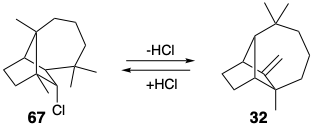
Para ilustrar las trampas de diseñar una síntesis molecular compleja, primero consideraremos algunas estrategias infructuosas para la síntesis de longifoleno. Una estrategia 4 se basó en la interconvertibilidad por reordenamiento del longifoleno (32) y su clorhidrato 67.
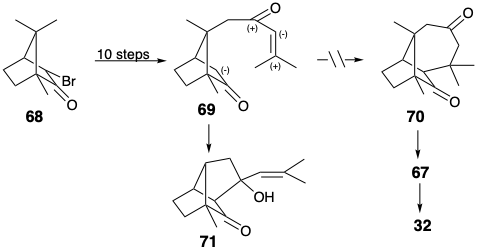
Se preparó un intermedio 69, relacionado con 65, en diez pasos a partir de D-α-bromoalcanfor (68), que está fácilmente disponible a partir de un producto natural. Sin embargo, 69 dio el producto aldólico 71 en lugar del producto deseado 70 en las condiciones de reacción de Michael. Así, a pesar de la disponibilidad inmediata del material de partida, la electrofilicidad ambiente del radical enona en 69 descarriló el plan sintético.
La eliminación de un enlace entre dos átomos no comunes en la primera dislocación de 32 condujo a la consideración del intermedio potencial 73 para la síntesis de longifoleno. 5 Esta ruta es especialmente atractiva ya que 73 se prepara fácilmente en pocos pasos a partir de materiales de partida fácilmente disponibles. La ciclación clave de 73 a 74 falló tras el tratamiento de 73 con ácidos.
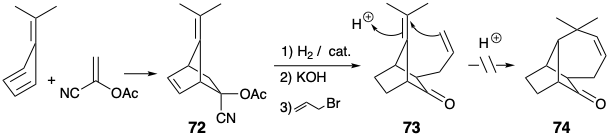
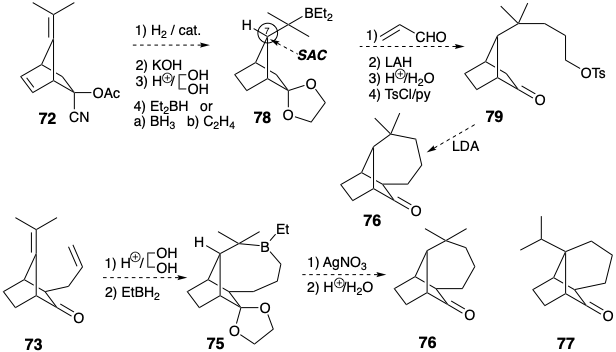
Deben examinarse otros modos de ciclación, como 73 → 75 → 76. Pero la hidroboración inicial preferencial del enlace C=C monosustituido excluirá la orientación requerida para la adición al enlace C=C tetrasstituido y conducirá a 77. Alternativamente, un intermedio 79, relacionado con 65, puede estar disponible en el aducto de Diels-Alder 72 y puede sufrir liberación de alquilación intramolecular 76. El control del enfoque estérico debe favorecer la estereoquímica requerida en la posición 7 en 78.
La primera síntesis exitosa de longifoleno (32) implica la ciclación clave 61 → 80 como último paso de la construcción esquelética. 3 Por cierto, 61 se sugiere no solo por consideraciones topológicas (es decir, estructura 55), sino también por análisis de reactividad polar (es decir, estructura 44). Después de mucha experimentación, solo se pudo lograr un rendimiento del 10-20% en este paso crucial. La conversión de 80 a 32 implicó entonces la adición final de un grupo metilo y metileno y la eliminación de los grupos carbonilo.
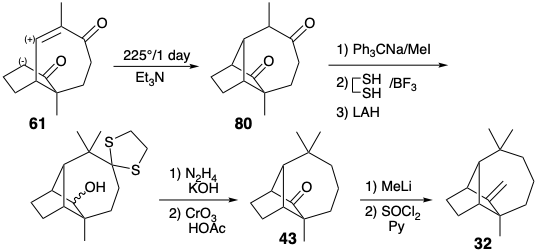
La síntesis del intermedio clave 61 ilustra una estrategia que es útil para la construcción esquelética de carbono, a saber, la modificación del tamaño del anillo (RSM). Así, se preparó 61 a partir de la cetona Wieland-Miescher fácilmente disponible (81) por expansión de un anillo de seis a siete miembros. La cetalización selectiva del grupo carbonilo saturado en 81 es posible debido a la desactivación del carbonilo insaturado por el sistema\(\pi\) de electrones adyacentes.
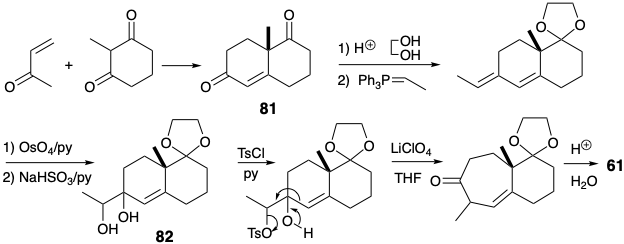
La exposición del diol 82 a las condiciones ácidas habituales para el reordenamiento pinacol-pinacolona resultaría en la ionización del alcohol alílico terciario y produciría un derivado de acetildecalina. Por lo tanto, fue necesario idear un procedimiento modificado para dirigir el reordenamiento del diol 82 a lo largo de la ruta deseada facilitando la ionización del hidroxilo secundario. Por lo tanto, el hidroxilo secundario se tosiló selectivamente. La ionización del grupo lábil de tosilato se acompañó de migración del grupo vinil. La cadena carbonada saturada es menos propensa a migrar que la insaturada porque la participación de p-electrones es posible en esta última pero no en la primera reordenación.
La táctica de modificación del tamaño del anillo
La lógica de una ruta sintética puede ser utilizada como herramienta para idear una estrategia o, ex post facto, como marco para lograr una comprensión fundamental de una síntesis conocida. La decisión de emplear la modificación del tamaño del anillo en la síntesis anterior de 61 es una consecuencia lógica del análisis topológico y polar de esta diana. El análisis topológico sugiere la desconexión del sistema de anillos bicíclicos en enlaces a los carbonos de la cabeza de puente que son átomos comunes. La doble desconexión del anillo de siete miembros sugiere un precursor simétrico, una 2-metilciclohexan-1,3-diona 2-sustituida. El análisis polar revela la posibilidad de un recocido polar para la construcción de la ciclohexandiona que explota la activación proporcionada por dos grupos carbonilo consonantes. Sin embargo, una de las desconexiones deseadas de 61 se encuentra en un circuito disonante. La eliminación de un átomo de este circuito disonante (contracción del anillo) produce un circuito consonante en 82 y la posibilidad de construcción esquelética por annelación polar; es decir, la annelación Robinson produciendo 81.
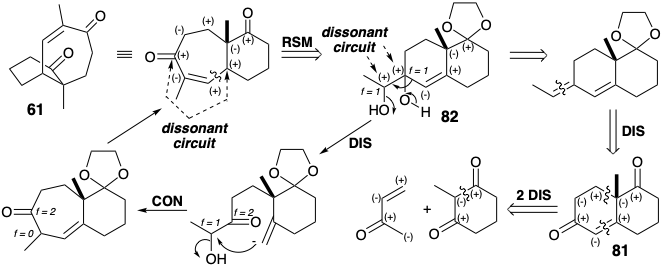
Es importante señalar que un circuito disonante en 61 se produce a partir de un precursor disonante 82. También, como se señaló en el capítulo anterior (ver sección 3.4), el reordenamiento de expansión del anillo de 82 equivale a una hipotética dislocación de dos etapas del objetivo, la desconexión seguida de la conexión. También es instructivo anotar los cambios en f s que acompañan al reordenamiento 82 → 61. La desconexión polar eleva f (de +1 a +2) para el centro electrófilo que experimenta desconexión polar desde 82 y baja f (de +1 a 0) para el centro electrófilo que experimenta conexión polar. La disonancia de reactividad polar requerida es creada por una reacción no polar, hidroxilación vecinal oxidativa de un alqueno (adición dioxidativa). Este alqueno es obviamente derivable de la diona 81 por olefinación selectiva de Wittig. 81 es totalmente consonante. Se puede construir mediante reacciones polares a partir de 2-metilciclohexan-1,3-diona y metil vinil cetona. Si no se hubiera conocido el proceso de anelación de Robinson y la cetona de Wieland-Miescher (81), el análisis retrosintético anterior habría llevado a su invención.
Comprobar si hay fallas
Habiendo ideado la estrategia anterior, es obligatorio aplicar el paso 4 del “Protocolo para el Diseño Sintético” esbozado en la página 23. Debemos examinar la estrategia en busca de posibles fallas. De hecho, se espera que el reordenamiento polar de 82 bajo catálisis ácida siga una ruta alternativa que implica la migración de hidruro a un ion carbenio terciario que se formaría más fácilmente que el ion carbenio secundario requerido. Por lo tanto, la estrategia se modificó para proporcionar activación selectiva del hidroxilo secundario. Así, la tosilación potenció su nucleofugacidad.
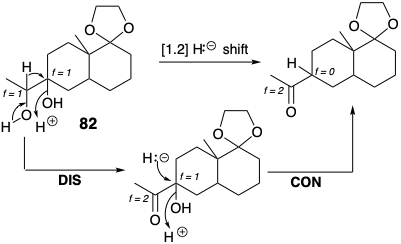
También tenga en cuenta que el reordenamiento migratorio concertado de hidruro es equivalente a una dislocación de dos etapas del objetivo, la desconexión seguida de la conexión de\({}^\ominus\) H. Además, la desconexión polar de hidruro eleva f (de +1 a +2) para el centro de carbono electrófilo que experimenta desconexión polar (el origen de migración) en 82 y disminuye f (de +1 a 0) para el centro de carbono electrófilo que experimenta conexión polar (terminal de migración).
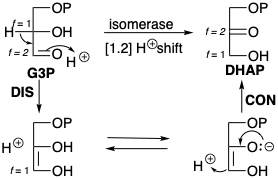
Cambios similares en f s acompañan a reordenamientos polares que involucran carbono nucleófilo en el origen y terminal de la migración como, por ejemplo, en el reordenamiento de G3P a DHAP (ver sección 2.1). Este proceso es en realidad una dislocación de dos etapas del DHAP objetivo: desconexión de H\({}^\oplus\) de C-1 seguida de conexión de H\({}^\oplus\) en C-2.
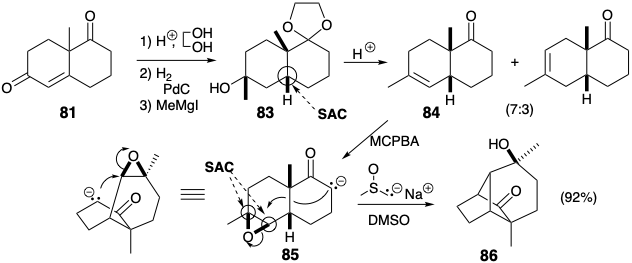
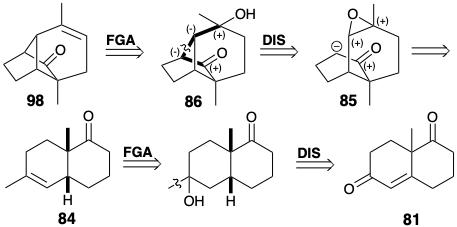
La modificación del tamaño del anillo se puede aplicar en cualquier etapa de la construcción esquelética. En la siguiente síntesis, la expansión del anillo se aplica después de completar una red esquelética que es topológicamente equivalente a la del longifoleno (32). 6 Aunque la red esquelética en 86 tiene puentes de diferentes longitudes que los de 32, tiene la misma conectividad que 32. La expansión de uno de los puentes en 86 conduce al sistema de anillos longifoleno (ver abajo). La síntesis de 86 tiene varias características importantes. Al igual que en la síntesis previa de 32, el presente appraoch comienza con la cetona de Wieland-Miescher (81). La hidrogenación catalítica procede con la formación estereoselectiva de la cis-decalona 83 debido a la adición controlada por aproximación estérica de hidrógeno al lado convexo del sistema de anillos plegados de 81. De igual manera, la epoxidación de 84 ocurre con la entrega estereoselectiva de oxígeno desde el lado convexo. La estereoquímica del epóxido 85 es ideal para el ataque nucleofílico durante la alquilación intramolecular S N 2 del anión enolato correspondiente. Esta ciclación clave en la síntesis de longifoleno de Mc Murray procede con un excelente rendimiento (92%).
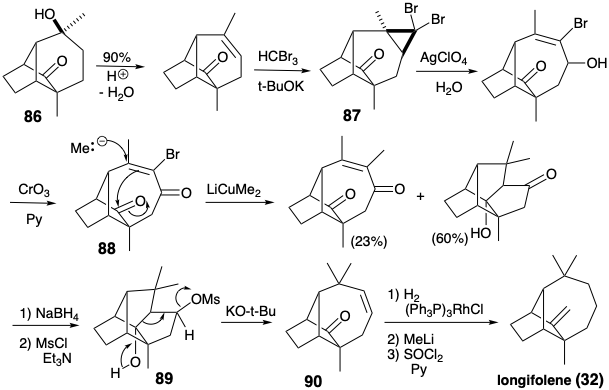
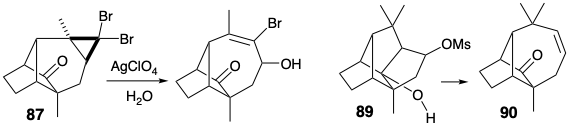
El sistema de anillos de longifoleno se generó a partir de 86 por una expansión de anillo que implicó la apertura pericíclica de un ion ciclopropil carbenio que se genera durante la solvolisis de 87. La adición nucleofílica requerida de un nucleófilo de metilo a una enona 88 estuvo acompañada de dos reacciones no deseadas. Una, la sustitución de un sustituyente vinil-bromo por metilo, generó un subproducto inútil. Sin embargo, el otro, una condensación intramolecular de aldol, no fue un defecto fatal porque el anillo extra así formado podría ser segmentado por una reacción de fragmentación (89 → 90).
Expansión de anillo como un proceso de tres pasos
Nuevamente, hagamos un análisis retrosintético ex post facto para lograr una comprensión más fundamental de la síntesis de longifoleno a través de intermedios clave 81 - 90. Consideraremos algunas alternativas que no fueron adoptadas, y examinaremos consideraciones estratégicas que subyacen al camino que se eligió. En este análisis, presumiremos las condiciones límite de usar 81 como material de partida y generar una red tricíclica de carbono mediante la formación de un enlace entre los átomos comunes incipientes 1 y 2 (numerados como en 55 en la página 116) en un precursor bicíclico. Además, la funcionalidad se introducirá presumiendo que una cetona es el progenitor del grupo metileno exocíclico. Sin embargo, en lugar de formar el esqueleto tricíclico al final de la síntesis después de la expansión de un anillo de 6 a 7 miembros, primero formaremos el esqueleto tricíclico y luego realizaremos una expansión del anillo. Podríamos presumir que el carbono cuaternario que lleva la gema dimetilos se inserta en el anillo de seis miembros de un precursor 92 para generar 91. Que 91 pueda contener un segundo carbonilo adyacente a la cabeza de puente es lo que sugiere el hecho de que este carbono en 92 corresponde a un carbono carbonilo en el material de partida 81 (vide infra). El enlace a desconectarse entre dos átomos comunes en 92 se encuentra en un circuito disonante entre los carbonilos. Por lo tanto, se requiere funcionalidad adicional, es decir, un nucleófugo, en un precursor, X en 93, para permitir la formación de enlaces polares.
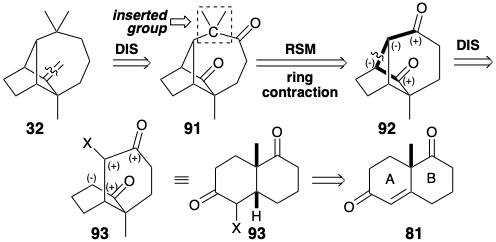
La expansión del anillo implica la inserción de un átomo de carbono entre dos miembros del anillo. Se debe formar un enlace entre el carbono nuevo y cada miembro del anillo, mientras que el enlace entre los miembros del anillo debe ser cortado. Hay dos formas topológicamente diferentes de lograr una expansión de anillo. Una posibilidad para generar 91 a partir de 92 es análoga a la expansión del anillo de 81 vía 82 (ver arriba). Así, se logra una dislocación retro pinacol de 91 desconectando el carbono cabeza de puente (como nucleófugo) en 91 del carbono cuaternario y reconectándolo (como nucleófilo) al carbono carbonilo vecino. Esto sugiere un sintón 94 y un equivalente sintético 95 como precursores de 91. En esta estrategia, la expansión del anillo se logra mediante una secuencia de conexión-desconexión-conexión (CDC) que inicia con la conexión del carbono nucleofílico del 2-diazopropano a un carbono carbonílico electrófilo de 92.
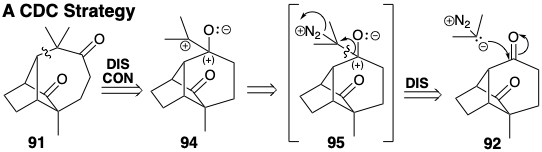
Una estrategia topológicamente diferente, conexión-conexión-desconexión (CCD), implica necesariamente un intermedio de ciclopropano que podría formarse por cicloadición a un alqueno 98. Así, 91 podría derivarse de un ciclopropano 97 que podría isomerizarse a un precursor de ciclohepteno 96. Necesariamente, solo uno de los grupos gema metilo del 91 puede estar presente en 96 porque el carbono que porta este metilo es cuaternario en el precursor ciclopropilo 97. Por lo tanto, se debe prever la introducción del último grupo metilo. Esto podría hacerse agregando funcionalidad a 96, como en 99 que tiene un grupo carbonilo conjugado con el centro de carbono al que se debe agregar un metilo. Si la expansión del anillo que producirá 99 implica una fragmentación polar del enlace de fusión del anillo en un intermedio de ciclopropano, entonces el análisis polar retrosintético sugiere dos rutas a 99. En ambas vías, el enlace de fusión del anillo es proporcionado por la conexión retrosintética a una funcionalidad relacionada con subdiana electropofílica portadora de carbono, un carbonilo en 99 o un hidroxilo en 100. Los electrones para esta conexión son proporcionados por un nucleófugo incipiente (Nu) a través de la adición al enlace C=C.
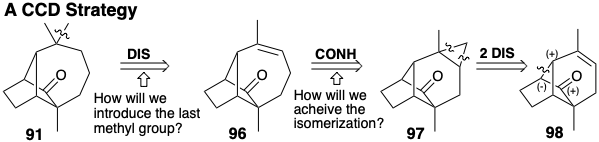
La ruta menos directa vía 100 es compatible con un precursor alqueno 98. Ambas vías reveladas por este análisis implican el cicloaditon de un carbeno al que se le añade un nucleófugo (Nu). Aunque la salida del nucleófugo podría ocurrir después de la fragmentación del enlace anillo-fusión, es posible un tiempo alternativo. La solvolisis del dibromociclopropano derivado del 98 probablemente sería un proceso concertado.
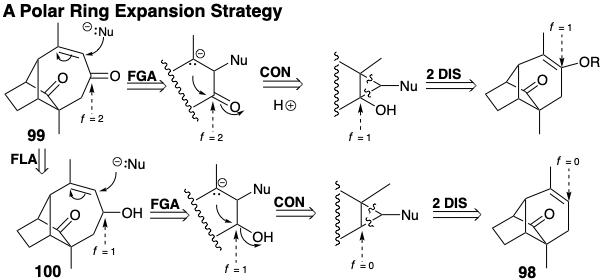
La dislocación polar de 98 a un electrófilo alílico y enolato puede proporcionar una estrategia defectuosa porque el enlace C=C en 99 introduce electrofilicidad ambiente. Así, la ciclación podría generar 102 por una reacción de S N 2'.
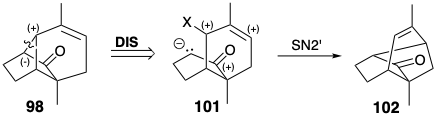
Por lo tanto, el enlace C=C en 98 se introduce mejor después de la ciclación, por ejemplo, por deshidratación del alcohol 86. El enlace a desconectarse entre dos átomos comunes en 86 se encuentra en un circuito disonante entre los grupos carbonilo e hidroxilo. Por lo tanto, se requiere funcionalidad adicional en un precursor, por ejemplo 85, para permitir la formación de enlaces polares. Este epóxido sería obtenible por adición dioxidativa a un alqueno 84. La generación de 84 a partir de la cetona de partida 81 es trivial.
Fragmentación de Bicíclicos Fusionados: Una Táctica para Generar Anillos Más Grandes
Durante la síntesis de Mc Murray de longifoleno, una conexión indeseada formada por condensación intramolecular aldólica del enolato 103 generó en la adición conjugada de un nucleófilo de metilo al intermedio 88. Debido a la proclividad del enolato 105 hacia la condensación aldólica, la fragmentación retro aldólica del producto pentacíclico 104 no pudo proporcionar el sistema de anillos requerido. Este problema fue eludido por una fragmentación isoelectrónica (ver página 80) luego de disminuir el nivel de funcionalidad de la cetona en 104 a un alcohol. Así, la fragmentación retro Prins del mesilato 89 generó 90 en los que el alqueno débilmente nucleofílico, en contraste con el enolato nucleofílico más fuertemente en 105, no mostró proclividad hacia la condensación con un grupo carbonilo.
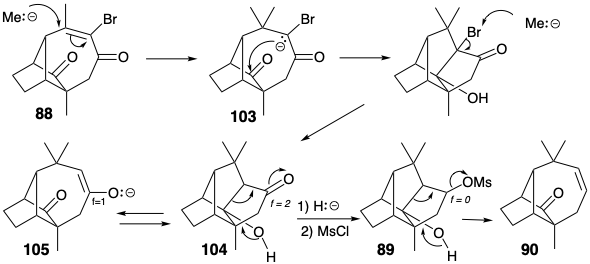
La síntesis de Mc Murray de longifoleno proporciona dos ejemplos de fragmentación de un enlace compartido por dos anillos fusionados para generar un solo anillo más grande. El primer ejemplo aprovechó la fragmentación del ciclopropano 87 como parte de una táctica de expansión de anillo, mientras que el segundo, un paso no planificado en la síntesis, implicó la fragmentación de 89.
Otra síntesis de longifoleno fue diseñada para explotar la fragmentación de un ciclobutano fusionado. Esta estrategia reconoce la posibilidad de usar funcionalidad carbonilo en 43 para proporcionar reactividad polar para introducir los grupos metileno y a-metilo, y otro grupo carbonilo para permitir la introducción de la matriz de gema dimetilo en un precursor 106. La dislocación de esta subdiana por una conexión polar sugiere que la diona 106 podría ser generada por la fragmentación retroaldólica de una β-hidroxicetona 107. En contraste con el equilibrio entre el aldol 104 y la diona 109 que favorece al primero, se espera que el equilibrio entre el aldol 107 y la diona 106 favorezca a la segunda debido al alivio de la tensión anular asociada a la escisión de un ciclobutano.
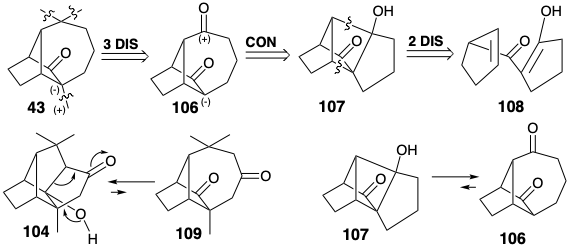
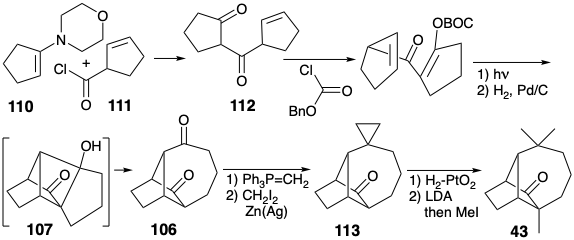
Una síntesis excepcionalmente eficiente de longifoleno resultó de la aplicación de esta estrategia. 7a Solo se utilizan 10 etapas para convertir la enamina 110 y el haluro de acilo 111 en longifoleno con un rendimiento global de 26%. La fototólisis de un derivado éster de enol de la diona 112 seguida por la eliminación hidrogenolítica del grupo bencioxicarbonil (BOC) generó la diona 106 vía 107. La metilenación selectiva del carbonilo menos congestionado estéricamente en 106 seguido de ciclopropanación, hidrogenólisis de 1 1 3 y metilación liberó la cetona 43, un intermedio en las síntesis de longifoleno de Corey y McMurray.
Una ruta de ciclación de polienos
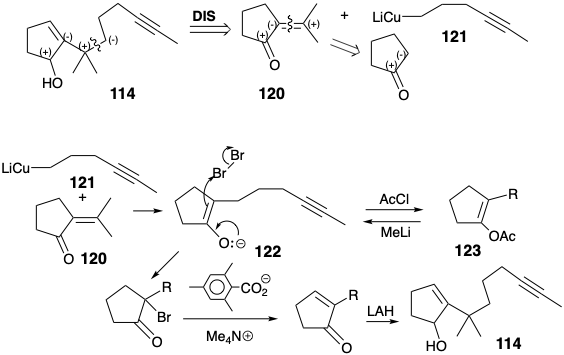
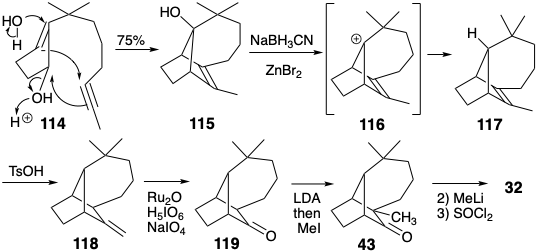
Otra síntesis eficiente del longifoleno (32) se basa en una simplificación estructural sugerida por el análisis topológico. Así, la dislocación a una subdiana 60 (ver página 116) por la eliminación de dos enlaces que involucran átomos comunes sugiere un precursor que contiene solo un anillo. En la síntesis, estos dos enlaces se generaron en una ciclación clave de polieno catalizada por ácido (114 → 115). 7b La conversión de 115 a 32 requiere la eliminación reductora del hidroxilo. Esto se logró mediante un reemplazo de S N 1 de hidroxilo por hidruro a través de un ion carbenio intermedio 116. Para proporcionar activación polar que pudiera explotarse para introducir el grupo metilo angular, el enlace C=C en 117 se isomerizó a un metileno exocíclico en 118. La escisión oxidativa luego entregó la cetona 119.
Se sugiere una síntesis de 114 a partir de un electrófilo de metilenociclopentanona 120 y un sintón nucleofílico de cadena lateral 121 mediante análisis polar. Se agregaron dos etapas adicionales a la síntesis para permitir la purificación del enolato 122 producido por la adición 1,4-de 120 a 121. Así, 122 quedó atrapado por O-acilación. Después de la purificación del acetato de enol 123, el enolato 122 se regeneró y luego se bromó. La deshidrobromación y reducción completó la síntesis de 114.