4.5: Homo y Bishomo Sesquiterpenos ii Cecropía Hormonas Juveniles
- Page ID
- 70302

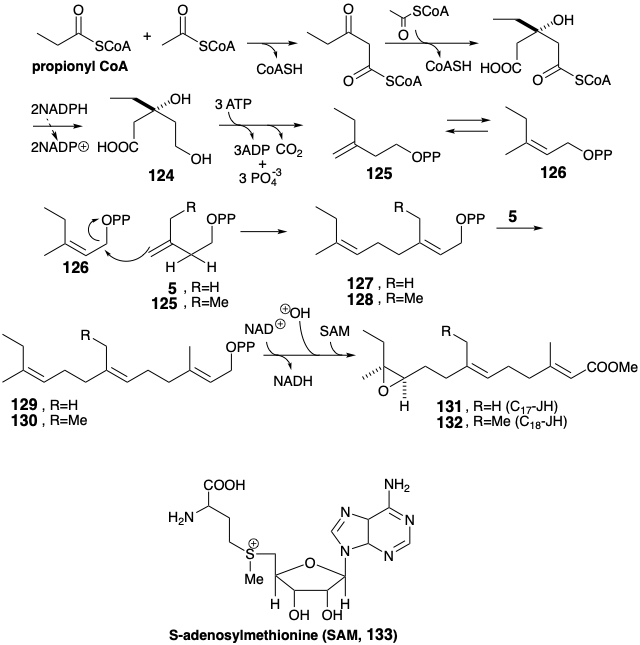
Las hormonas juveniles de Cecropia son parientes cercanos biogenéticos del farnesol que contienen uno o dos átomos de carbono adicionales en sus esqueletos de carbono sesquiterpenoides. Estos homólogos terpenoides se denominan sesquiterpenos homo y bishomo respectivamente. Los átomos de carbono adicionales surgen a través de la incorporación de una o dos moléculas de propionato en lugar de acetato durante una construcción esquelética biosintética que por lo demás es idéntica a la del farnesol. Así, propionil CoA se condensa con dos moléculas de acetil CoA para dar ácido homomevalónico (124) después de la reducción con NADPH. La conversión de 124 a 125 mediante eliminación descarboxilativa e isomerización a 126 es seguida de la adición del electrófilo alílico 126 al enlace C=C terminal en Δ 3-isopentenil-PP (5) o a su homólogo de seis carbonos ( 125). Los electrófilos alílicos 127 o 128 luego alquilan Δ 3-isopentenil-pirofosfato para entregar homo y bishomo farnesilpirofosfatos 129 o 130, respectivamente. Estos se oxidan a los ácidos correspondientes por donación de hidruro a NAD+. La epoxidación enantio-selectiva y O-metilación del carboxilo por transferencia de metilo de S-adenosil-metionina (SAM, 133) da epoxiésteres ópticamente activos 131 y 132 que se conocen como hormonas juveniles C 17 y C 18 respectivamente.
Generación Estereocontrolada de Alquenos Trisustituidos
El desafío sintético primario que presentan las hormonas juveniles de Cecropia es el estereocontrol durante la construcción de dobles enlaces C=C trisustituidos. Los anillos temporales se pueden utilizar eficazmente para controlar la geometría del alqueno. Se han empleado diversas aplicaciones de esta táctica para lograr síntesis estereocontrolada de hormonas juveniles. Estas síntesis generalmente implican la construcción estereocontrolada de bishomo farnesoato de metilo (134), que se convierte en C 18-JH (132) por epoxidación regioselectiva como en la biosíntesis de 132. El análisis polar de 134 sugiere una síntesis a partir del carbanión 136 estabilizado con fosfono-éster y la dienona 135.
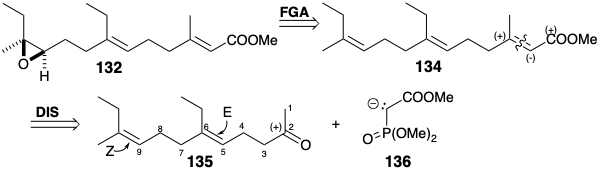
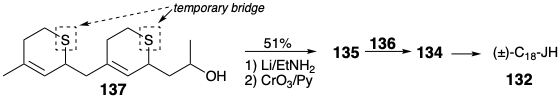
Una estrategia de puente temporal
Una táctica para la síntesis estereocontrolada de 135 emplea puentes tioéter temporales en 137 para hacer cumplir las configuraciones 5-E y 9-Z.8 Los puentes de azufre se escinden reductivamente después de completar el esqueleto de carbono de 135. Ambos fragmentos de dihidrotiapirano en 137 fueron derivados de un intermedio común, tetrahidro-1,4-tiapirona (138). El azufre cumple un doble papel estratégico en este esquema. Además de imponer la configuración requerida en los enlaces p carbono-carbono (estereocontrol), el azufre estabiliza un centro nucleofílico vecino (control de reactividad) en los carbaniones 139 y 140. Los grupos funcionales mercapto y sulfuro proporcionan fácilmente activación para reactividad nucleófila o electrófila. Son grupos funcionales bifílicos y, como se encontró anteriormente para el grupo funcional nitrilo bifílico (ver páginas 19 y 31), pueden ser utilizados para producir inversión de reactividad polar. Este fenómeno se encontró previamente en la conversión de un carbonilo electrófilo en un carbanión ditiano nucleofílico, un equivalente de carbanión acilo.
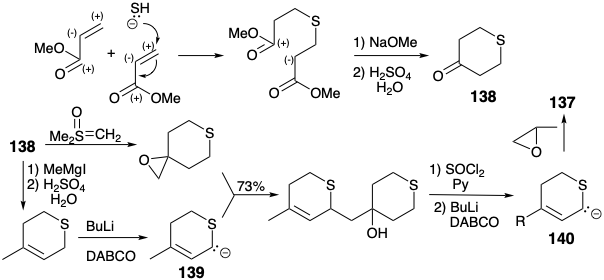
En la presente síntesis de JH, el β-carbono electrófilo del acrilato de metilo es transformado por el grupo funcional tioéter en un centro nucleófilo en 139 y 140.
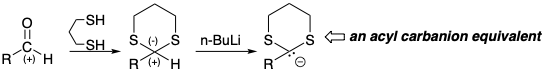
Otro caso de activación bifílica por azufre se encuentra en la reacción del carbono carbonílico electrófilo de 138 con metilida de dimetiloxosulfonio (141), un nucleófilo estabilizado con azufre, para producir epóxido 143. El azufre luego proporciona activación electrófila en el mismo carbono al servir como nucleófugo en la betaína intermedia 142.
SEC en Fragmentaciones
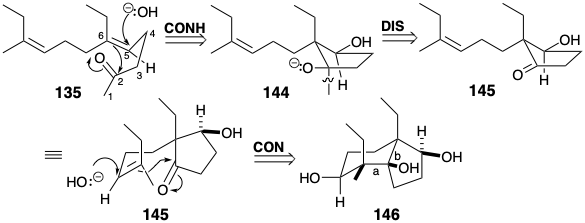
Otra síntesis estereocontrolada de 135 explota la adición estereoespecífica reversible, antiperiplanar a un enlace π carbono-carbono para transponer las relaciones estereoquímicas en intermedios monocíclicos y bicíclicos a las de productos acíclicos. 9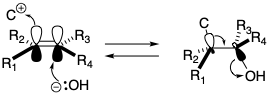 La disposición antiperiplanar permite solapamiento continuo durante una fragmentación concertada. Así, la configuración E del enlace 5,6-π-en 135 se conserva en forma latente en el intermedio cíclico 144 por una dislocación que implica la adición antiperiplanar de un electrófilo de carbono y un nucleófilo hidroxilo a 1 3 5. Un nuevo carbono carbonílico electrófilo es generado por la dislocación 144 fi 145. La configuración Z del enlace C=C en 145 se conserva en forma latente por una dislocación que implica la adición antiperiplanar de un electrófilo de carbono y un nucleófilo hidroxilo al enlace C=C en 145. Así, toda la información estereoquímica en el intermedio acíclico 135 está contenida en forma latente en el precursor bicíclico 146. El control de la geometría de alqueno en un esqueleto de carbono acíclico se transpone de este modo para controlar la estereoquímica relativa en una red de carbono multicíclica. Dado que las conformaciones de las redes de carbono multicíclicas son más rígidas que las acíclicas, las influencias de los efectos estéricos y de grupos vecinos son más fáciles de predecir y generalmente más pronunciadas.
La disposición antiperiplanar permite solapamiento continuo durante una fragmentación concertada. Así, la configuración E del enlace 5,6-π-en 135 se conserva en forma latente en el intermedio cíclico 144 por una dislocación que implica la adición antiperiplanar de un electrófilo de carbono y un nucleófilo hidroxilo a 1 3 5. Un nuevo carbono carbonílico electrófilo es generado por la dislocación 144 fi 145. La configuración Z del enlace C=C en 145 se conserva en forma latente por una dislocación que implica la adición antiperiplanar de un electrófilo de carbono y un nucleófilo hidroxilo al enlace C=C en 145. Así, toda la información estereoquímica en el intermedio acíclico 135 está contenida en forma latente en el precursor bicíclico 146. El control de la geometría de alqueno en un esqueleto de carbono acíclico se transpone de este modo para controlar la estereoquímica relativa en una red de carbono multicíclica. Dado que las conformaciones de las redes de carbono multicíclicas son más rígidas que las acíclicas, las influencias de los efectos estéricos y de grupos vecinos son más fáciles de predecir y generalmente más pronunciadas.
La fragmentación de los enlaces a y b en 146 se promovió mediante la conversión de hidroxilo en grupos salientes de toluenosulfonato. El triol 146 fue fácilmente monotosilado selectivamente al menos hidroxilo secundario estéricamente congestionado. La fragmentación del alcohol b-tosiloxi 3° resultante que suministra el Z-alqueno 145 procedió estereoespecíficamente tras el tratamiento con\(\ce{NaH}\). La adición del\(\ce{MeLi}\) derivado tetrahidropiranilo de 145 ocurrió estereoselectivamente (57%). Después de la desprotección y tosilación selectiva del hidroxilo secundario en el diol resultante, la fragmentación de un alcohol intermedio β-tosiloxi 3° ocurrió suavemente para proporcionar 135 estereoespecíficamente (80%).
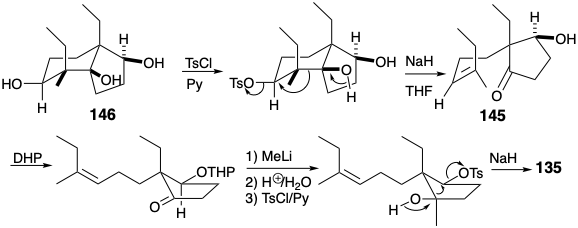
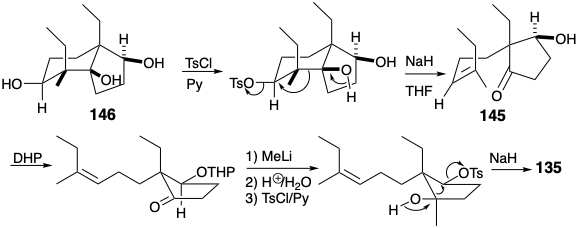
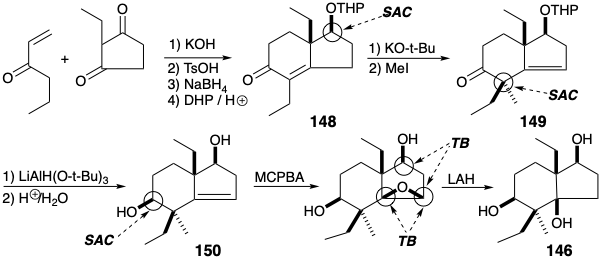
Dado que la funcionalidad en 146 fue introducida por reacciones polares, es una conclusión perdida que los grupos funcionales están conectados por circuitos consonantes. El análisis polar de 146 muestra que todos los circuitos en el anillo de ciclohexano son consonantes. Esto permite la desconexión de enlaces a los átomos comunes de fusión del anillo. Sin embargo, la activación de la reactividad nucleofílica vecinal a los hidroxilos secundarios requiere una conjugación que solo es proporcionada por grupos carbonilo como en 147. La desconexión polar de 147 luego sugiere propil vinil cetona y 2-etilciclopentan-1,3-diona como materiales de partida.
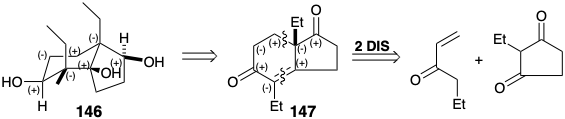
Una síntesis de 146 basada en la estrategia descrita anteriormente comenzó con la anelación Robinson de 2-etilciclopentano-1,3-diona con propil vinil cetona. El enfoque estérico controló el suministro de hidruro a la diona resultante proporcionó 148 después de la protección del hidroxilo. El control del enfoque estérico también resultó en metilación estereoselectiva de la cara α menos congestionada estéricamente del enolato de 148. De manera similar, el suministro de hidruro a la cara menos congestionada estéricamente de 1 4 9 dio 1 5 0 estereoselectivamente. La estereoquímica del grupo hidroxilo final requerido para el triol 146 estuvo dictada por el efecto del hidroxilo vecino en 150 sobre la epoxidación de este alqueno. El hidrógeno del grupo hidroxilo se une con MCPBA, lo que impone el suministro de oxígeno cis al hidroxilo vecino como para 250 en la sección 3.6.
SEC a través de conformaciones preferidas
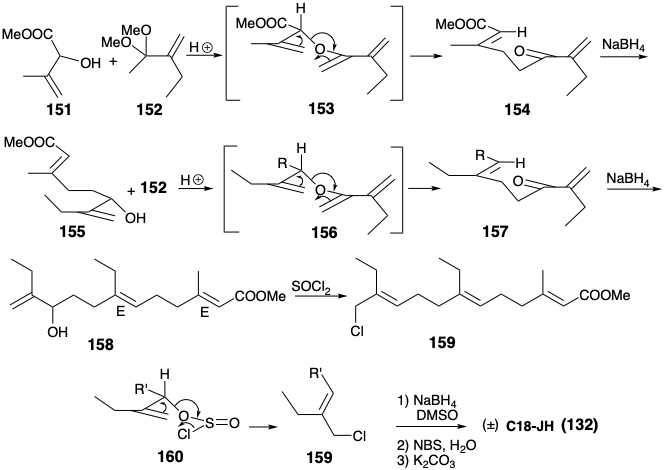
Los efectos conformacionales en estados de transición cíclicos también pueden resultar en estereoselectividad sustancial, así como predecible, en la generación de alquenos acíclicos. En otra síntesis estereocontrolada de C 18-JH (132), el reordenamiento de Claisen del éter alílico vinílico 153 a partir de la transcetalización de 152 con 151, seguido de la eliminación, genera estereoselectivamente la cetona γ, δ-insaturada 154. 10 Se prefiere un estado de transición silla con un sustituyente carbometoxilo ecuatorial para este reordenamiento sigmatropico [3.3]. La reducción de borohidruro de 154 da un alcohol alílico 155, el cual fue nuevamente homologado estereoselectivamente con 152 para dar alcohol alílico 158 vía 156 y 157. Un estado de transición cíclica es también la clave para una conversión estereocontrolada de 158 al cloruro 159 transpuesto alílicamente. Así, el éster de clorosulfito 160 de 158 da 159 vía transposición S N i' que implica una conformación de silla con el sustituyente voluminoso en una posición ecuatorial.
A continuación se describe otra síntesis de C 18-JH (132) que explota un reordenamiento de Claisen para generar estereoselectivamente el doble enlace 6,7-trisustituido. 11 Un paso interesante en esta síntesis es la destrucción selectiva de un subproducto no deseado 162. Este cloruro alílico es más reactivo que los isómeros 161 y 163 y, por lo tanto, forma selectivamente una sal de piridinio soluble en agua. El reordenamiento de claisen ocurre a través de una conformación 164 en estado de transición con un grupo cloroetilo ecuatorial. El doble enlace 2,3-se genera estereoselectivamente por cis-1,4-adición de un organocuprato derivado de 165 a 2-butinoato de metilo.
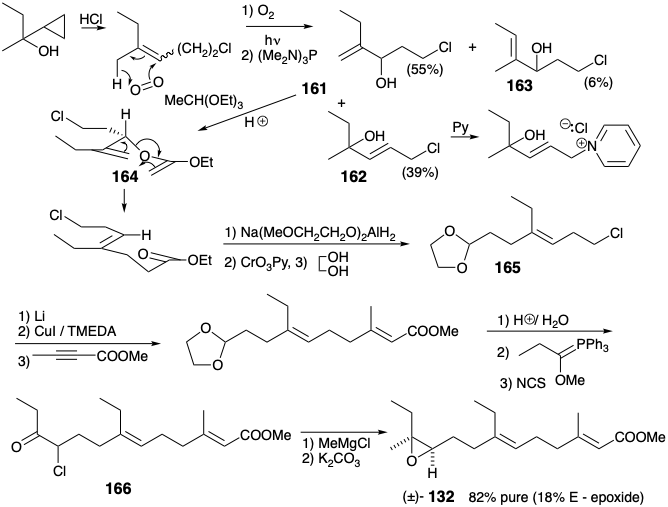
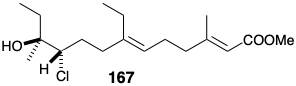
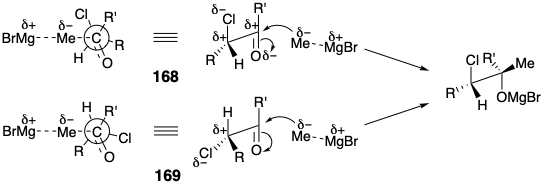
En esta síntesis, el epóxido se produce por ciclación inducida por bases de una clorohidrina en lugar de epoxidación de una olefina. La generación estereoselectiva de HO del Z-epóxido es posible porque la reacción de la clorocetona 166 con cloruro de metil magnesio conduce predominantemente a un diastereómero, la treo clorhidrina 167.
Esta estereoselectividad no es consecuencia de un estado de transición cíclica. Más bien, para las cetonas acíclicas que contienen sustituyentes α polares (por ejemplo, halógenos) que es poco probable que se coordinen con átomos metálicos, se debe considerar una combinación de tensión torsional, interacciones estéricas e interacciones electrostáticas. Se han formulado dos modelos para explicar dicha estereoselectividad. Un modelo presume que la conformación reactiva de tales cetonas es una estructura (por ejemplo, 168) en la que el grupo carbonilo y el sustituyente α polar son antiperiplanares para minimizar la repulsión dipolo-dipolo. Alternativamente, el estado de transición puede parecerse al 169 que permite la separación máxima del sustituyente electronegativo a y el reactivo nucleofílico cargado negativamente. Otro ejemplo de dicha estereoselección controlada estereoelectrónicamente lo proporciona la conversión 48 + 49 → 53 presentada en el Capítulo 3 (sección 3.3).
Otras estrategias de TB
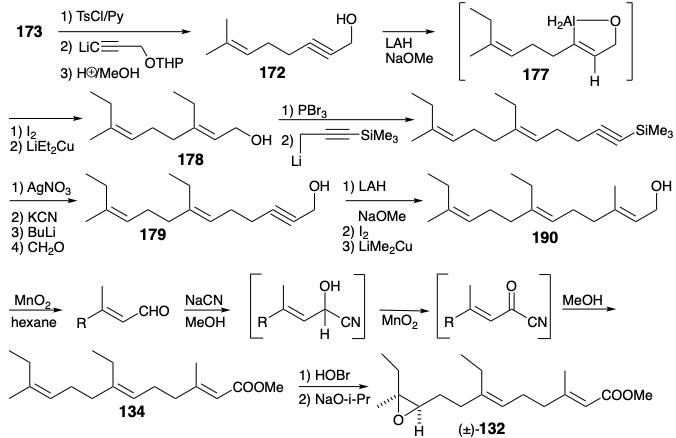
Dos tácticas para la construcción estereoselectiva de enlaces C=C trisustituidos no encontrados en los ejemplos anteriores se explotan en una estrategia de síntesis de C 18-JH que fue ideada por Corey. 12 La configuración 2,3-E en 134 se puede asegurar mediante un anillo temporal durante la hidroaluminación pseudo-intramolecular de un alcohol propargílico 171 que produce un vinil alano intermedio 170 el cual es posteriormente alquilado. Esta táctica también puede producir la configuración 6,7-E mediante una secuencia similar de hidroaluminación-alquilación aplicada a 172. El hecho de que un alquino terminal sea un carbanión latente sugiere una dislocación polar de 172 a un nucleófilo acetilido derivado de alcohol propargílico y un precursor electrófilo 173. La configuración E de 173 se puede asegurar mediante un puente temporal que se sugiere por dislocación a un precursor 174 más altamente funcionalizado con grupos carbonilo diferenciados. Así, el acoplamiento reductor de estos dos grupos carbonilo proporciona un precursor 175 de dicarbonilo latente puenteado.
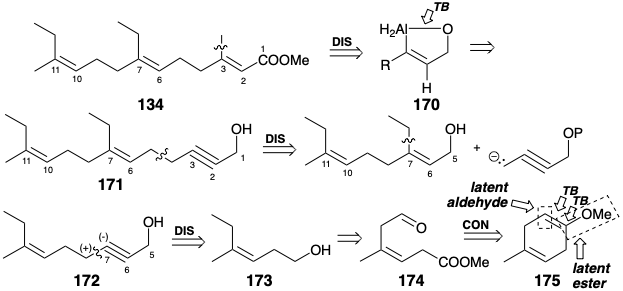
Destaca especialmente el papel sintético del grupo aldehído carbonilo en 174. Este grupo funcional es explotado únicamente para facilitar el estereocontrol.
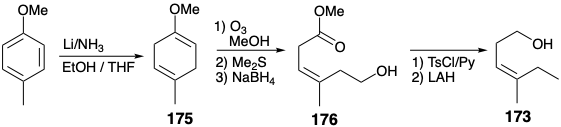
Los sustituyentes en el producto acíclico 176, de la escisión oxidativa selectiva de 175, son necesariamente cis ya que solo un doble enlace cis puede acomodarse en un anillo de seis miembros. La relación cis entre el sustituyente aluminio e hidroximetilo forzada en el intermedio 177 por un puente temporal se conserva en la halogenólisis posterior del enlace C-Al y la alquilación del yoduro de vinilo resultante que suministra 178. Ambas transformaciones ocurren con la retención de la configuración. Una secuencia similar entrega 180 estereoselectivamente de 179. La conversión del alcohol alílico 180 en el intermedio clave 134 requiere oxidación seguida de O-metilación.
SEC a través de conformaciones preferidas
La formación altamente estereoselectiva de alquenos trisustituidos a veces se puede lograr mediante procesos que no involucran intermedios con puentes temporales o estados de transición cíclicos. Así, ocasionalmente, una combinación de efectos conformacionales y estereoelectrónicos puede producir alta estereoselectividad en reacciones de moléculas acíclicas. Por ejemplo, el enlace 6,7-C=C de JH se puede generar estereoselectivamente durante la transformación del bromuro de ciclopropilcarbinilo 181 en el bromuro homoalílico 182. 13
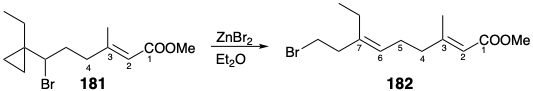
La estereoselectividad surge de una preferencia estereoelectrónica por una disposición antiperiplanar de enlaces de escisión que conduce a la generación del nuevo doble enlace carbono-carbono de una manera concertada y estereocontrolada. Es decir, una disposición coplanar de los enlaces de ruptura C-C y C-Br permite el acoplamiento de estas escisiones de enlaces con la formación de dobles enlaces carbono-carbono. Cualquiera de las conformaciones 183 o 184 del estado de transición satisface este requisito, pero 183 se prefiere claramente porque el grupo ciclopropilo eclipsa solo el hidrógeno. Así, un sesgo conformacional, aunado a una preferencia estereoelectrónica, favorece un estado de transición 183, que conduce a la E-olefina 182.
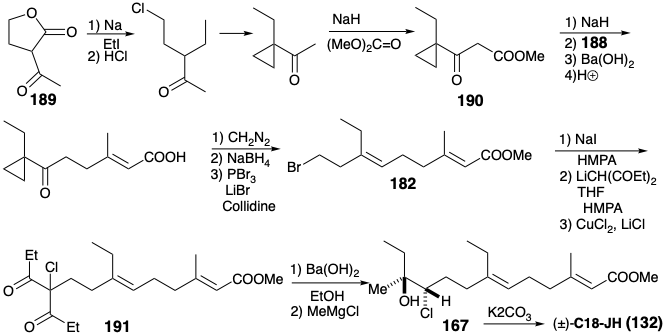
La preparación de un intermedio temprano, 188, para el segmento C1 a C4 de 181 ilustra una táctica útil para la síntesis de estereoisómeros puros: destrucción selectiva de uno de dos productos isoméricos a partir de una reacción no estereoselectiva. Así, la N-bromosuccinimida bromina el ácido dimetacrílico (185) de forma no selectiva para dar una mezcla de bromoácidos 186 y 187.
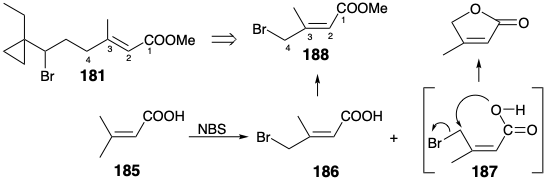
El isómero Z no deseado 187 se somete a lactonización espontánea, dejando el isómero E deseado 186 como el único producto orgánico ácido, que puede extraerse en base suave y posteriormente metilarse para proporcionar el intermedio 188 para la síntesis de JH. Esto se combina con un nucleófilo derivado del cetoéster 190 que está disponible, a su vez, de 1-acetil-γ-burirolactona 189. La simetría se aprovechó durante la finalización del esqueleto de carbono JH por alquilación del enolato de 3,5-heptanodiona con 182. La cloración del producto dio 191. El grupo extra propionilo se escindió en una reacción retro de Claisen por\(\ce{Ba(OH)2}\). La clorocetona 166 resultante reaccionó estereoselectivamente con MeMgCl para dar 167 con menos de 8% del diastereómero no deseado (ver sección 4.6). La heterociclización inducida por bases de la treo clorohidrina 167 suministró C 18-JH racémico. 13