
5.3: Tetraciclinas
- Page ID
- 70276

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
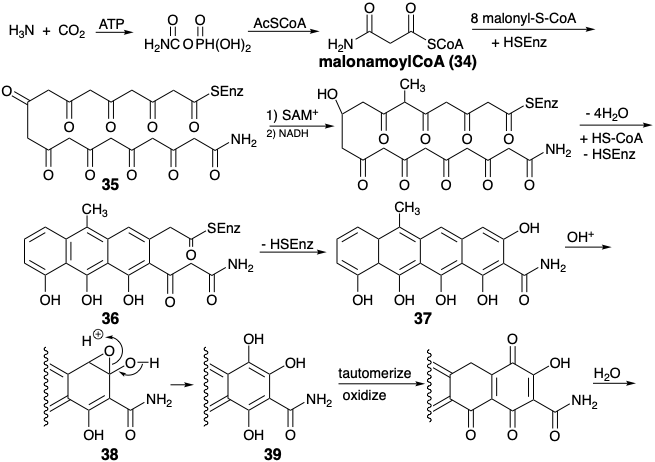
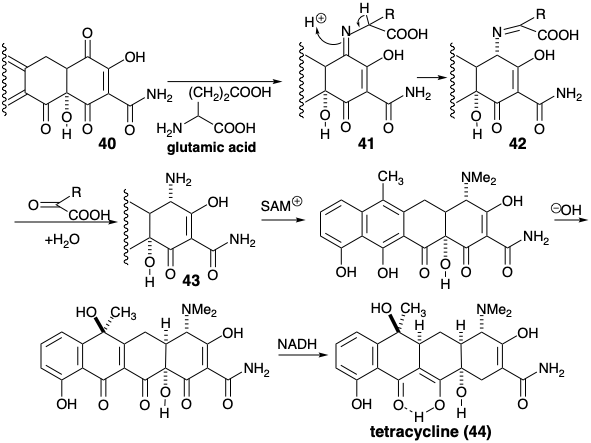
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)El acetil CoA no es el único tioéster que puede iniciar la oligomerización de la matriz enzimática de malonil CoA. Así, por ejemplo, se ha postulado una biosíntesis para la tetraciclina (44) que implica la condensación de malonamoil CoA (34) con ocho moléculas de malonil CoA para producir un tioéster de policetoamida unido a enzima 35, que se deshidrocicla parcialmente después de la metilación de uno metileno y reducción de un carbonilo. 6 El anillo final del sistema de anillos de tetraciclina se forma por ciclación de Dieckmann de 36 a 37 después de la liberación del policétido parcialmente ciclado de la policétido sintetasa. Dos hidroxilaciones aromáticas aumentan la funcionalidad después de completar el esqueleto de carbono. Estas hidroxilaciones pueden involucrar óxidos de areno intermedios, por ejemplo 37 → 38 → 39. La aminación reductora de 40 vía 41 y 42 para producir 43 se acompaña de desaminación oxidativa del ácido glutámico.
Análisis topológico de sistemas de anillos fusionados. 7
Un par de anillos es un sistema de anillos fusionados si dos anillos comparten uno y solo un enlace común, el enlace de fusión. Los sistemas de anillo de esteroides y tetraciclina son ejemplos de estructuras multicíclicas que contienen solo pares de anillos fusionados, a diferencia de los pares espirocíclicos o de anillos puenteados. Además de los enlaces de fusión (marcados con f), los diagramas a continuación indican átomos comunes (mostrados como •), y enlaces exendo (marcados e) que son exo a un anillo y endo a otro, y enlaces (indicados como líneas discontinuas) que se forman durante la biosíntesis de estos sistemas de anillos a partir de precursores acíclicos.
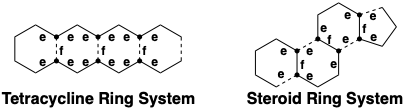
Existe un interesante contraste entre las estrategias topológicas de la biosíntesis de tetraciclina y esteroides. Ambas estrategias involucran intermedios acíclicos clave que incorporan todos los átomos de carbono esqueléticos. Sin embargo, la biosíntesis del esqueleto de tetraciclina implica la formación de todos los enlaces de fusión de anillos, mientras que solo se forman enlaces exendo durante la biosíntesis del esqueleto esteroideo. Otro contraste se encuentra en la generación biosintética del anillo periférico, es decir, el anillo que permanece después de la escisión de todos los enlaces de fusión. Así, solo se genera un enlace periférico durante la biosíntesis del esqueleto de tetraciclina, mientras que durante la biosíntesis de esteroides se generan cuatro enlaces del anillo periférico.
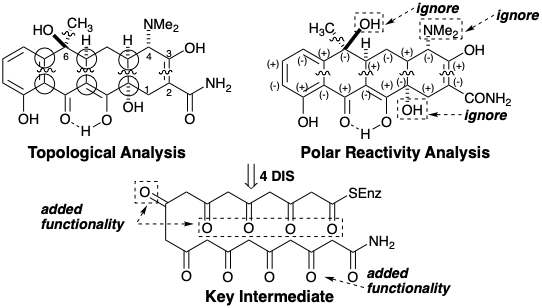
También es digno de mención el hecho de que, en ambas biosíntesis, los enlaces entre pares de átomos comunes son enlaces estratégicos, es decir, estratégicamente importantes para lograr una rápida reducción de la complejidad molecular por desconexión durante la dislocación de la diana sintética. Las dislocaciones de la estrategia biosintética de tetraciclina se recomiendan tanto por análisis de reactividad topológica como polar. Topológicamente, la estrategia desconecta todos los enlaces entre pares de átomos comunes. El análisis de reactividad polar revela una amplia funcionalidad. Si se ignora la activación proporcionada por varios grupos funcionales, numerosos grupos funcionales permanecen con circuitos de conexión únicamente consonantes que pueden generarse explotando la funcionalidad relacionada con la diana en precursores junto con varios grupos funcionales consonantes agregados (carbonilo).
Una estrategia lineal para tetraciclinas
La primera síntesis de un derivado de tetraciclina biológicamente activo (aunque no natural) fue lograda por Woodward y colaboradores. Estos trabajadores simplificaron el objetivo sintético al no incluir el 6-hidroxilo terciario lábil así como el 6-metilo de la tetraciclina (44). El anillo A de 45 es funcional y estereoquímicamente la porción más compleja de esta diana simplificada. Este anillo está tan altamente funcionalizado que el análisis polar es ambiguo. Contiene una plétora de disonancias de reactividad polar. Si este anillo se corta de 45, resulta una tremenda simplificación del objetivo sintético. No sólo se elimina la abundancia de funcionalidad reactiva, sino que también se realiza una simplificación topológica. Así, al escindir un par de enlaces exendo que son vecinales y cocíclicos (en el mismo anillo primario, es decir, uno que no es diseccionado en un par de anillos más pequeños por un puente transanular), se eliminan todos los vestigios del anillo A. El sintón BCD restante es un fragmento relativamente estable químicamente, estructuralmente simple 46. Un posible intermedio sintético, es decir, fragmento molecular apropiadamente funcionalizado, que corresponde a 46 es 47. El grupo carbonilo en el anillo B en 47 proporciona activación para la elaboración del anillo A. El éter metílico en el anillo D bloquea la desprotonación del fenol. El análisis polar de la porción no aromática de 47 sugiere una dislocación a 48 y dimetiloxalato, un biselectrofilo disonante simétrico. La anelación de 48 por acilación Friedel-Crafts de 49 explotaría la reactividad nucleofílica del anillo D aromático en 49, que es activado por un grupo metoxi relacionado con la diana, y la reactividad electrófila de un carbonilo.
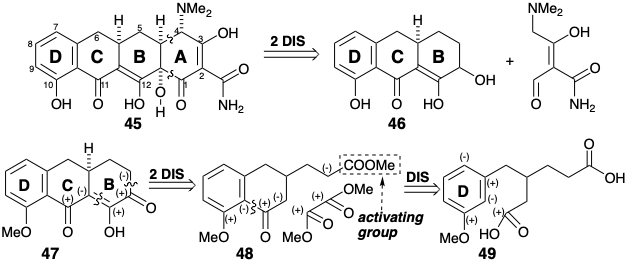
Sin embargo, esta estrategia es potencialmente defectuosa debido a que la sustitución electrófila que debe ocurrir orto con respecto al sustituyente metoxi en el anillo D de 49 durante la conversión a 48, también podría ocurrir en la posición nucleófila para al grupo metoxi activador. Para impedir la paracilación, se podría usar un sustituyente cloro en el precursor 50 como grupo bloqueante (un sustituyente introducido para controlar la reactividad y posteriormente eliminado).
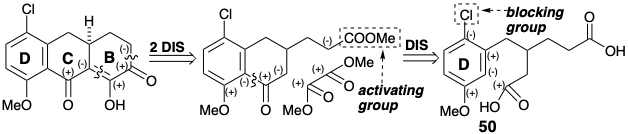
Topológicamente, es deseable una dislocación de 49 a 51 + 52 porque todos los vestigios de la cadena lateral se eliminan del anillo aromático. Sin embargo, el análisis polar de 49 muestra que la alquilación o acilación electrófila de un precursor de anisol favorecería la sustitución orto o para en lugar de la meta sustitución requerida para generar 49.
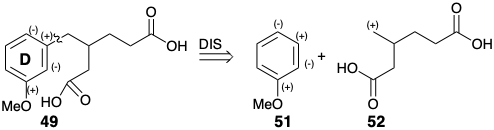
La dislocación 49 ⇒ 53 + 54 es recomendada por la disponibilidad inmediata de organometálicos bencílicos correspondientes a 53. Un sintón nucleofílico como 53 podría proporcionar un diéster de 49 por 1,4-adición a 54. De hecho, se conoce una síntesis general de ácidos alcanodioicos β-sustituidos como 49 que se relaciona con este enfoque. 9 La dislocación a intermedios β-cetoéster cíclicos más conectados 55 revela la posible utilidad de precursores de cicloalquenona fácilmente disponibles para la síntesis de ácidos alcanodioicos β-sustituidos por escisión retro de Dieckman.
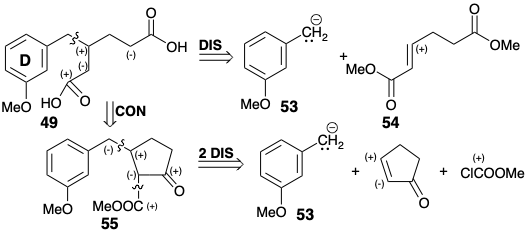
En la práctica, la C-acilación de los intermedios enolatos como el 56 se acompaña de O-acilación de los β-ceto-ésteres resultantes. Sin embargo, no se requieren etapas adicionales debido a que los ésteres de enol resultantes se hidrolizan a β-ceto-ésteres bajo las condiciones de reacción requeridas para la escisión retro Dieckman de estos últimos para generar los ácidos alcano dioicos β-sustituidos diana.
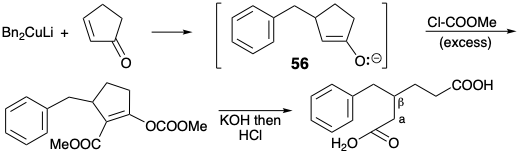
En lugar de un nucleófilo bencílico como material de partida, la estrategia de Woodward se basó en la elección de un electrófilo bencílico fácilmente disponible, metil m-anisato (57), como material de partida. Esta elección canaliza el análisis retrosintético a un precursor 58 con un grupo funcional activador, el carbonilo bencílico, que debe eliminarse posteriormente para proporcionar 49. Aunque la conjugación con el carbometoxilo remoto en 58 podría proporcionar la activación nucleofílica requerida para unir un material de partida de éster hexanodioico con 57, Woodward optó por explotar un método sintético clásico para cetonas, alquilación de un β-ceto-éster seguido de hidrólisis y descarboxilación (una síntesis de éster acetoacético), para ensamblar el esqueleto de carbono de 49. Esta elección exige la inclusión de un grupo activador de éster carboxílico en 58.
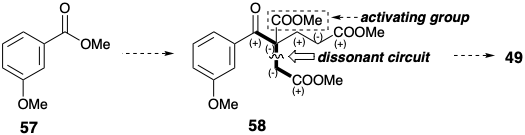
Woodward examinó tres estrategias diferentes para descubrir qué ruta realmente proporciona los mejores rendimientos generales de 58 de 57. Cada ruta aprovecha los materiales de partida fácilmente disponibles. Curiosamente, la ruta más larga (menos convergente), usando acetato de metilo y cloroacetato de metilo como bloques de construcción, dio mejores rendimientos globales que las otras dos rutas que explotan precursores de diésteres simétricos. También es significativo que, en cada ruta, se utilice un material de partida disonante para proporcionar el circuito disonante en 58.
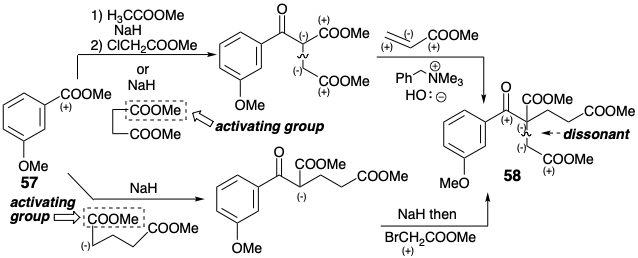
La conversión de 58 al intermedio tricíclico clave 61 se logró luego por hidrólisis, descarboxilación, hidrogenólisis, cloración, acilación de Friedel-Crafts, ciclación de Claisen-Condensación-Dieckmann, y luego otra hidrólisis y descarboxilación. La interesante desmetilación selectiva que produjo 60 resultados de la transesterificación intramolecular de un alcohol bencílico intermedio seguido de hidrogenólisis del éster bencílico resultante. La metilación de 60 se realizó únicamente para facilitar la purificación.
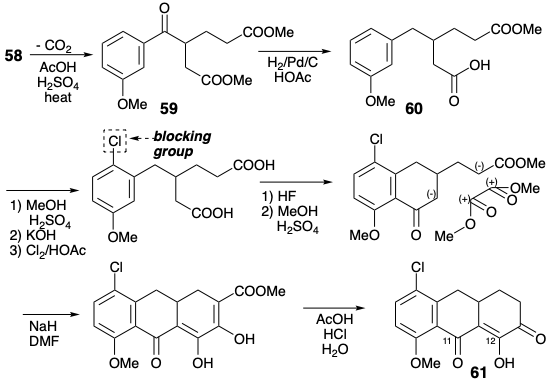
Woodward anticipó diferentes propiedades para los tres grupos carbonilo en 61. Así, la matriz β-dicarbonilo (C-11 y 12) es “un sistema estabilizado de ácido carboxílico vinilógico, mientras que el tercero, al igual que en los simples α-cetoácidos, debe ser tanto altamente susceptible a las reacciones de adición como fácilmente enolizable. Esta última propiedad debe conferir una alta reactividad nucleofílica al metileno adyacente”. Esta reactividad se aprovechó para agregar a 61 un fragmento precursor para el anillo A, y luego el grupo dimetilamino por reacciones polares. El tercer carbonilo, habiendo cumplido su propósito, se eliminó luego por reducción a α-hidroxicetona 63, activación por transesterificación intramolecular y escisión reductora de la lactona resultante.
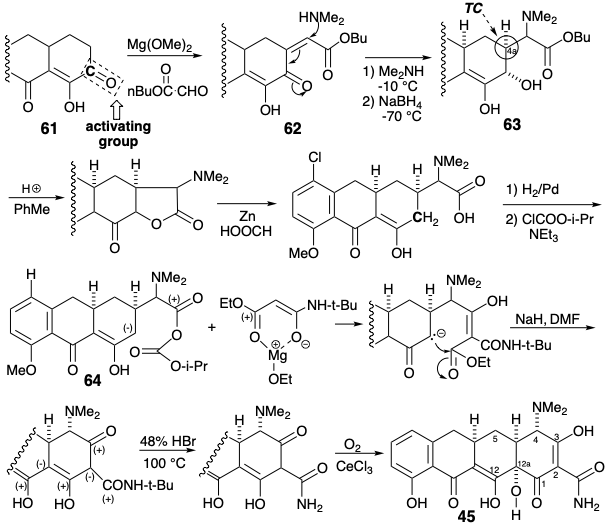
Luego se elaboró un precursor acíclico completamente funcionalizado para el anillo A por acilación de un carbanión de malonamato de metilo con el anhidrido mixto 64. La ciclación de dieckman, aprovechando la reactividad nucleofílica conferida al metileno adyacente por el carbonilo en la posición 12, produjo el anillo A. La estereoquímica requerida en la posición 4a en 64 se produce durante la adición de dimetilamina a 62, lo que favorece la más estable epímero ecuatorial de 63. La instalación del último grupo funcional, el hidroxilo en la posición 12a se realizó entonces oxidativamente para entregar 45.
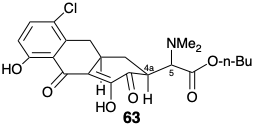
Una estrategia más convergente para las tetraciclinas.
En otra estrategia para la síntesis total de 6-desmetil-6-desoxitetraciclina (45), como en la síntesis previa de Woodward, el objetivo se simplifica eliminando el hidroxilo C-12a. Entonces, a excepción de la activación polar proporcionada por el sustituyente C-4 dimetilamino, la reactividad polar proporcionada por los grupos funcionales restantes es totalmente consonante a lo largo de cualquier circuito como se muestra en 65. La desconexión polar 65 ⇒ 66 separa la molécula en dos grandes fragmentos unidos por un simple puente de metileno. Otras desconexiones de enlace desconectan el anillo A en dos precursores de cadena recta 67 y 68, que pueden reunirse mediante reacciones polares. Los equivalentes sintéticos que Muxfeldt utilizó para los sintones 67 y 68 fueron 69 y 70a, respectivamente. 9
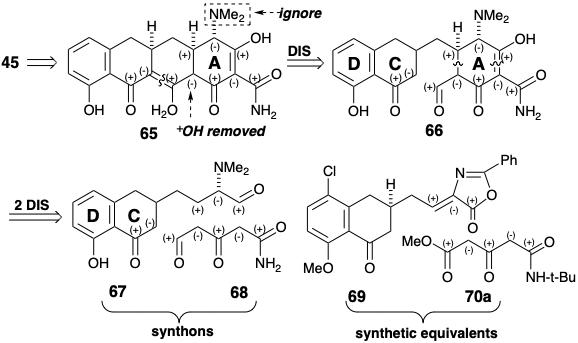
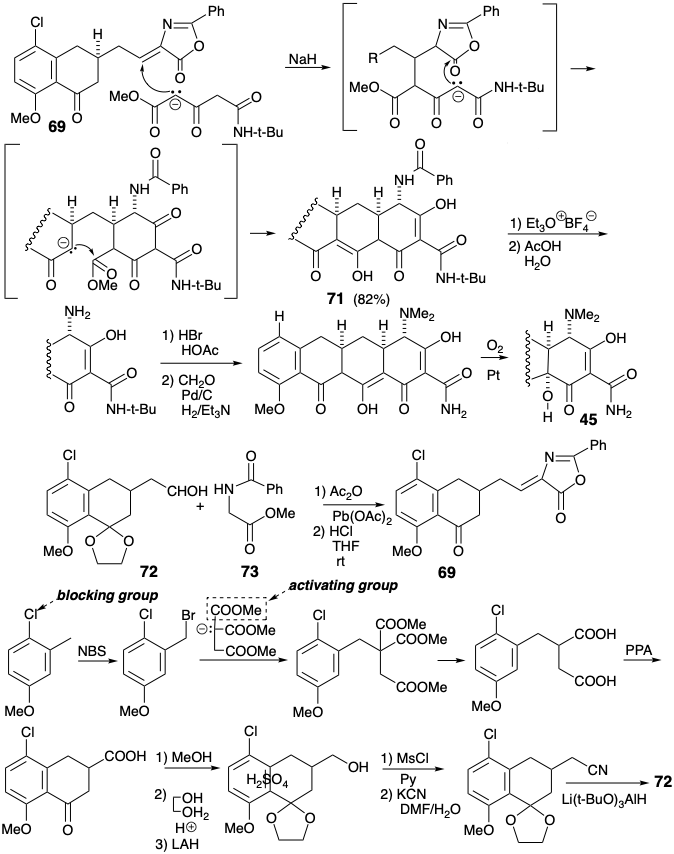
Lo más destacado de la síntesis de Muxfeldt es la ingeniosa reacción que produce el intermedio tetracíclico 71 en un solo paso a partir del precursor bicíclico 69 ¡en 82% de rendimiento! El ajuste final de la funcionalidad permite fácilmente 45 de 71. La estrategia se beneficia de un alto grado de convergencia. Además, 69 está fácilmente disponible por una síntesis de azalactona de 72 y 73. Finalmente, se preparó el aldehído 72 de anillo de CD intermedio con buen rendimiento global a partir de 4-cloro-3-metilanisol.
La primera síntesis total de una tetraciclina natural, el derivado 5-hidroxi terramicina, fue lograda por Muxfeldt. 10 Los anillos A y B se ensamblaron por la misma estrategia utilizada anteriormente para generar 45. Así, un aldehído 74 de anillo de CD, análogo al 72, se condensó con una azatio-lactona 75 preformada análoga a una azalactona intermedia implicada en la reacción de 73 con 72. El aceptor de Michael resultante 76, análogo al 69, se condensó luego con la amida desprotegida 70b correspondiente a 70a para generar el sistema de anillos de tetraciclina en una etapa sintética. La posterior desprotección de los hidroxilos enmascarados y la introducción oxidativa del último hidroxilo en la posición 12a fue seguida por la eliminación del grupo enmascarante tibenzoílo en condiciones excepcionalmente suaves tras el tratamiento con yoduro de metilo. Esta reacción implica la generación y posterior hidrólisis de un intermedio de éster metiltioimídico. Finalmente, la dimetilación controlada de la amina primaria dio terramicina.
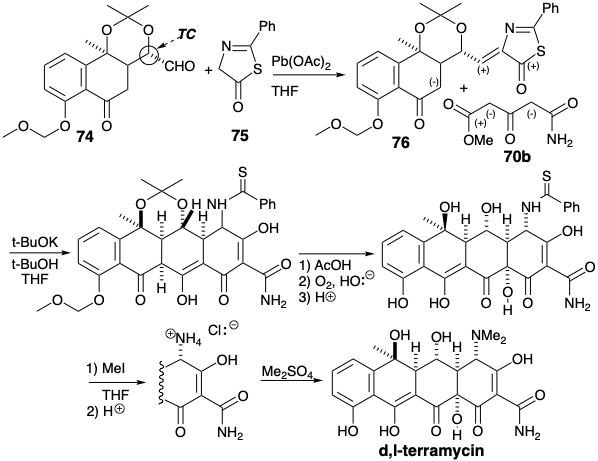
La estrategia empleada para generar el intermedio clave 74 explota un puente temporal en 78 para enmascarar el aldehído en forma latente como un alqueno y para crear un precursor tricíclico plegado que se espera que agregue un nucleófilo de metilo al carbonilo en la posición 6 desde el mínimo cara convexa estérica congestionada. Esto asegura una relación cis entre el grupo metilo y el protón cabeza de puente vecino. Se espera que la matriz vinílogosa α-dicetona en 77 sea especialmente electrófila, lo suficientemente activada para evitar la competencia por la adición al acetato carbonilo. La selectividad que favorece la adición al carbonilo en la posición 6 en lugar de 11 puede ser el resultado de una disminución de la electrofilicidad debido a la conjugación del 11- pero no el 6-carbonilo con el oxígeno en la posición 10. La presencia de un ciclohexeno en 77 recomienda una síntesis de cicloadición que involucra un dienófilo de quinona doblemente activado deficiente en electrones y 1-acetoxi-1,3-butadieno relativamente rico en electrones.
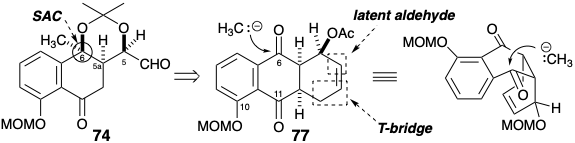
De hecho, la reacción de Diels-Alder de acetoxibutadieno con acetato de juglona (78) dio el diacetato 79 regioselectivamente, y la adición de un reactivo de Grignard de metilo a 79 es regio- y estereoselectiva. El anillo de ciclohexeno, habiendo cumplido su propósito, se escindió oxidativamente para generar un aldehído a partir de su equivalente latente, el doble enlace carbono-carbono. Un fragmento innecesario de dos carbonos fue luego eliminado por una secuencia que involucra condensación aldólica, escisión oxidativa y finalmente escisión retro de Claisen de una matriz intermedia de β-dicetonas en 80.
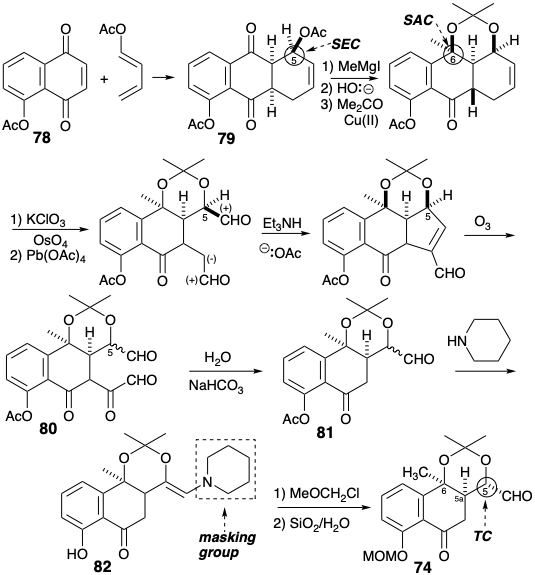
Debido a una preferencia estereoelectrónica por un estado de transición endo en la reacción de Diels-Alder de 78, 79 se genera con la configuración correcta en la posición 5. Sin embargo, la escisión oxidativa que genera 80 produce una mezcla de epímeros en la posición 5. Durante el reemplazo del grupo protector acetilo con un metoximetilo, se usa piperidina como nucleófilo para escindir el acetato y como grupo enmascarante para ocultar el aldehído sensible durante la alquilación del fenol. Afortunadamente, la hidrólisis de la enamina 82, por tratamiento con gel de sílice húmedo, generó 74 epiméricamente puro con la configuración requerida en la posición 5.