5.4: Eritronólido B
- Page ID
- 70293

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Una reacción promovida por enzimas de propionilCoA con metilmalonilCoA genera 83 enantioselectivamente. La reducción diastereoselectiva luego entrega el b-hidroxi tioéster 84. En contraste con la biosíntesis de ácidos grasos, la deshidratación y la reducción conjugada del tioéster α, β-insaturado resultante no precedió a la condensación de Claisen con un segundo equivalente de metilmalonilCoA durante la biosíntesis del precursor ácido seco 87 del macrólido 6-desoxieritronólido B.
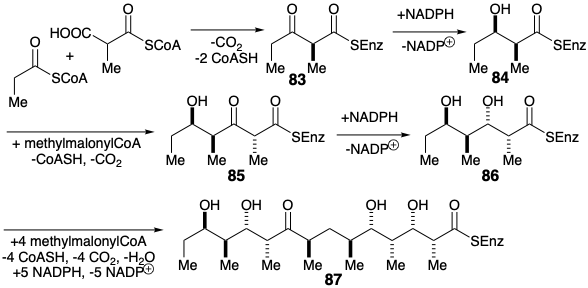
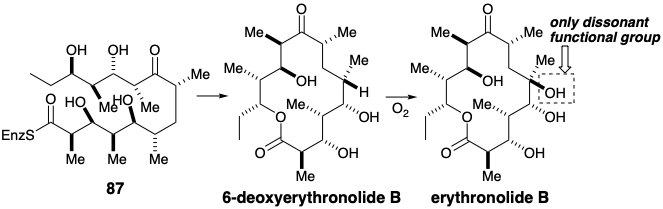
El β-cetotioéster resultante 85 se reduce a un β-hidroxiéster 86 que reacciona con metilmalonilCoA y NADPH adicionales para generar 87. La lactonización proporciona entonces 6-desoxieritronólido B a partir del cual se forma eritronólido por oxidación. La generación de solo 1 de los 2048 posibles diastereómeros del intermedio acíclico 87 es la notable consecuencia de la asimetría de los catalizadores homoquirales (enzimas) que promueven las condensaciones y reducciones responsables de producir diez centros asimétricos.
Una estrategia dirigida por relés para la síntesis total de eritronólido B.
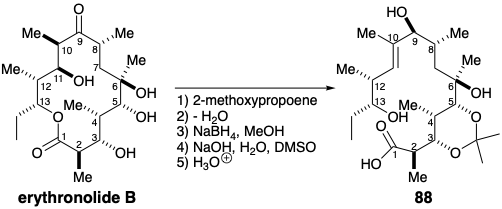
En un estudio de factibilidad, el eritronólido B de origen natural se convirtió en un hidroxiácido acíclico 88 que se utilizó para elaborar los pasos finales para una síntesis total. 12 El proceso de diseño sintético fue luego canalizado por la elección de este precursor de origen natural, un compuesto relevador. Los dos estereocentros en las posiciones 12 y 13 en 88 están alejados de los de las posiciones 2-6 y 8. Por lo tanto, sería difícil generar un conjunto de estereocentros bajo la influencia estéreo-controladora del otro. Más bien, los bloques de construcción que contienen estos estereocentros con las configuraciones absolutas correctas se pueden ensamblar y luego unir para proporcionar el compuesto de relé 88. La primera síntesis total de Eritronólido B1 3 adoptó una estrategia convergente asimétrica absoluta (ver sección 4.6) diseñada para proporcionar 88 mediante la unión de dos segmentos homoquirales, un nucleófilo 89 y un electrófilo 90.
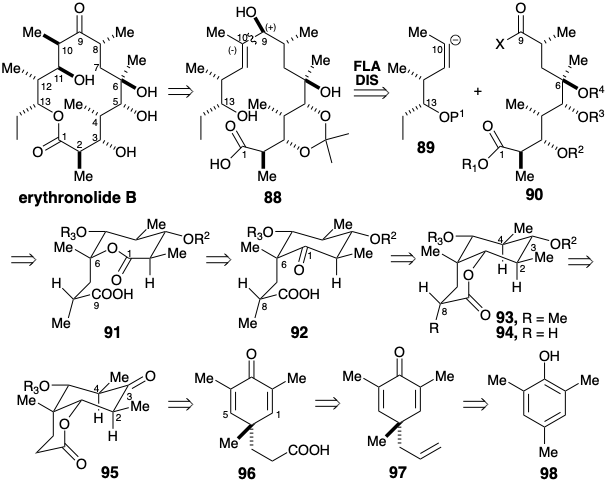
Para proporcionar una plataforma conformacionalmente rígida para generar y confirmar las configuraciones relativas de estereocentros, se concibió un precursor cíclico con puente temporal 91 para la subdiana acíclica 90. Aún mayor rigidez es proporcionada por el anillo más pequeño de una cetona 92 de seis miembros que incorpora la funcionalidad lactona en forma latente. Se puede esperar que el suministro axial de hidrógeno favorecido estereoelectrónicamente a la ciclohexanona isomérica 94 genere un hidroxilo ecuatorial en la posición 3. Además, la dislocación de 93 a 94 permite que un carbonilo en la posición 3 promueva una disposición ecuatorial termodinámicamente favorecida para los metilos en las posiciones 2 y 4, y prepara el escenario para descubrir un precursor simétrico, vide infra. Otro puente temporal, una lactona de seis miembros puede explotarse para favorecer la generación de la configuración requerida en el estereocentro relativamente remoto en la posición 8 en 92 durante una metilación de 95 que se puede esperar que favorezca un metilo ecuatorial en 94. Los puentes de lactona también pueden explotarse para asegurar la orientación estéreo y regioquímica adecuada durante la introducción de sustituyentes de oxígeno en las posiciones 1 y 6 mediante adiciones polares a los enlaces C=C en un precursor simétrico de dienona 96. Se puede usar una búsqueda en la literatura de materiales de partida disponibles con el esqueleto de carbono de 96 para identificar la alilciclohexadienona 97 que se prepara fácilmente a partir del trimetilfenol 98.
La reacción con bromuro de alilo de un fenolato de 98 proporciona 100 por reordenamiento de Claisen del producto inicial de O-alilación 99. Un reordenamiento Cope catalizado por ácido de 100 entrega la dienona simétrica 97 que se hidroborata selectivamente en el grupo vinilo terminal y luego se oxida para producir el ácido carboxílico 96. La administración estereoselectiva y regioselectiva de oxígeno a la posición 5 se logra mediante la adición intramolecular de un carboxilato a un enlace C=C en 96. La captura del intermedio de carbanión enolato formado reversiblemente con bromo electrófilo produce 101. Para repetir este proceso en el enlace C=C restante, la lactona se saponifica para regenerar un ácido carboxílico. Esto también genera un epóxido por desplazamiento intramolecular del sustituyente bromo por alcóxido. Una segunda entrega estereoselectiva y regioselectiva de oxígeno, esta vez a la posición 1, se logra nuevamente mediante la adición intramolecular de un carboxilato a un enlace C=C. La posterior eliminación reductiva de la funcionalidad heteroatómica innecesaria en las posiciones 1 y 4 proporciona 103.
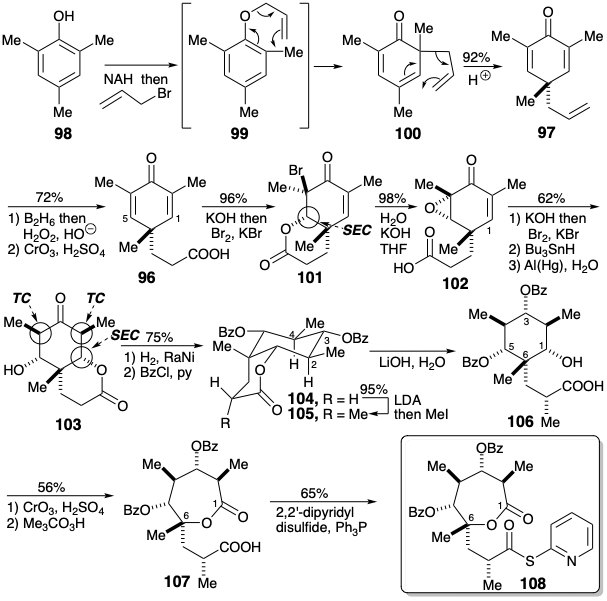
La activación nucleofílica proporcionada por la lactona carbonilo en 103 podría ahora explotarse para introducir un grupo metilo en la posición 8. Sin embargo, el ajuste previo del nivel de funcionalidad en la posición 3 evita la activación nucleofílica adyacente a la cetona carbonilo. La metilación de 104 luego proporcionó 105 estereoselectivamente. La lactona en 103 diferencia los sustituyentes hidroxilo en las posiciones 1 y 5. Para mantener esta diferenciación después de la saponificación de la lactona, los otros hidroxilos en 105 tuvieron que ser adecuadamente enmascarados. La elección del grupo enmascarante éster benzoato es particularmente sutil. La viabilidad de la saponificación selectiva de la lactona en presencia de ésteres de benzoato se basa en la disminución de la electrofilicidad del grupo benzoato carbonilo debido a la conjugación. La oxidación del alcohol 106 a una cetona y luego a una lactona 107 seguida de la activación del ácido carboxílico como tioéster proporcionó un segmento electrófilo C1-9 equivalente a 90 donde los grupos enmascarantes R1 y R4 se reemplazan por una lactona puente.
La síntesis esbozada anteriormente proporciona compuestos racémicos. La resolución de un intermedio temprano, el ácido carboxílico 102, por cristalización fraccionada de sales diastereoméricas de 1-α-naftiletilamina, se empleó para generar intermedios con las configuraciones absolutas mostradas anteriormente que se requieren para el eritronólido B natural.
También se empleó la resolución de un intermedio temprano para preparar el enantiómero requerido de un precursor 115 (ver sección 6.3) para el nucleófilo 89. Así, el ácido epoxi carboxílico 110, que está fácilmente disponible por una oxidación en un solo paso del alcohol trans - crotílico (109), se resolvió mediante cristalización fraccionada de sales diastereoméricas de 1-α-naftiletilamina. La sustitución nucleofílica estereoespecífica en 110 generó la configuración absoluta requerida en la posición 12. La regioselectividad de esta apertura de epóxido está controlada por el sustituyente éter voluminoso en 111. El uso del alcohol epoxi correspondiente en el desplazamiento mostró una regioselectividad muy inferior. El reemplazo del hidroxilo terminal en 112 por un grupo metilo para dar 113 se pudo lograr sin enmascarar el hidroxilo secundario usando un exceso de\(\ce{Me2CuLi}\). La conversión completamente regioselectiva del acetileno 114 en yoduro de vinilo 115 dependió de la regioselectividad sobresalientemente alta que se había reportado recientemente para la hidrocirconación de acetilenos asimétricamente disustituidos. Este paso en la síntesis de eritronólido B de Corey es un ejemplo conmovedor del impacto de los desarrollos en la metodología sintética en nuestra capacidad para lograr síntesis eficientes de moléculas orgánicas complejas.
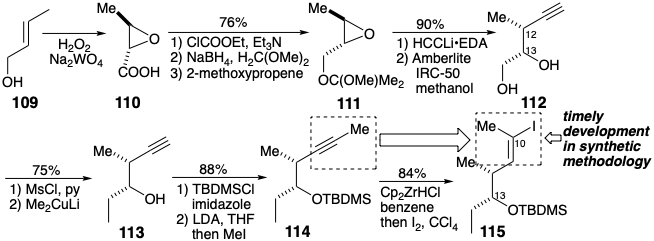
Aunque ambos bloques de construcción 108 y 115 estaban disponibles en forma homoquiral con las configuraciones absolutas correctas para eritronólido B, la síntesis se realizó en realidad acoplando el enantiómero correcto de 115 con 108 racémico. Así, un reactivo de Grignard 116 derivado del 115 se aciló con tioéster 108 para producir la cetona 117 y un diastereómero con 90% de rendimiento total. La mezcla se carió a través de varias etapas adicionales antes de la separación por cromatografía preparativa en capa fina.
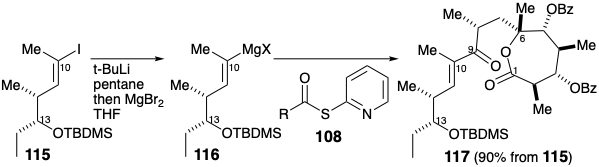
Así, la reducción de la cetona carbonilo en 117 resultó inesperadamente difícil debido a la sorprendentemente similar reactividad de los carbonilos cetónicos y lactona hacia la mayoría de los agentes reductores y también debido a una proclividad hacia la reducción conjugada de la enona. La reducción con borohidruro de zinc estuvo acompañada de dos fenómenos inesperados, una estereoselectividad completa muy bienvenida y una translactonización esencialmente irrelevante que generó una lactona 118 de 10 miembros después de la eliminación del grupo protector sililo en la posición 13. La saponificación de esta lactona se realizó de manera más efectiva con\(\ce{LiOH}\) una acuosa\(\ce{H2O2}\) que presumiblemente se beneficia de la supernucleofilia del anión hidroperóxido. La hidrólisis de los ésteres de benzoato menos reactivos en 119 se realizó luego con agua\(\ce{KOH}\). La metilación posterior suministró 120 junto con un diastereómero del cual se separó por TLC sobre gel de sílice.
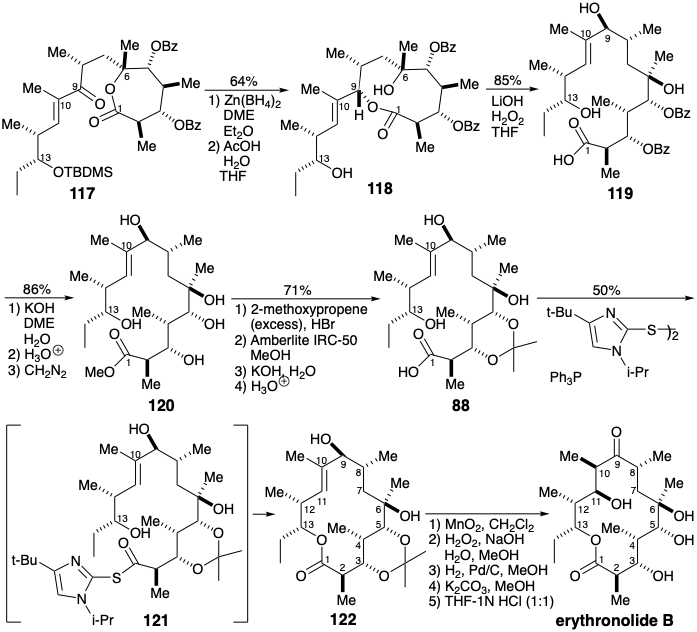
El compuesto relevador 88 se obtuvo a partir de 120 por cetalización con 2-metoxipropeno, hidrólisis selectiva de 2-metoxi-2-propil éteres que también se formaron, y saponificación del éster metílico. La macrolactonización se logró mediante el “método de doble activación” que implica la activación simultánea de las funciones hidroxilo y carboxilo. Presumiblemente, un intermedio 123 doblemente activado colapsa a un aducto carbonílico tetraédrico 124 del cual se forma la lactona 126 por eliminación de 125. Así, calentar el tioéster 122 a reflujo en tolueno seco proporcionó eritrololida B con 50% de rendimiento.
Eritronólido B a partir de Bloques de Construcción Homoquirales Derivados de Azúcar.
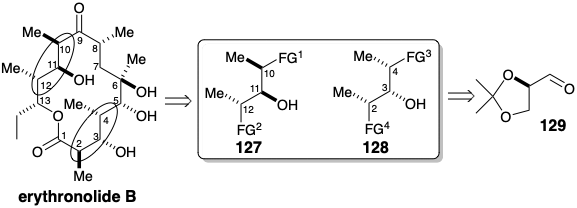
Una estrategia para una síntesis total enantioespecífica de eritronólido B evolucionó a partir del reconocimiento de que los segmentos C2-4 y C10-12 están sustituidos de manera idéntica pero tienen diferentes estereoquímicas absolutas. Dichos segmentos, diferencialmente sustituidos en cada extremo, es decir 127 y 128, podrían elaborarse y unirse para generar el producto natural. Por lo tanto, se lanzaron estudios para definir rutas sintéticas a dichos intermedios. Dado que el (R) -2,3-O-isopropilidenogliceraldehído (129) es un bloque de construcción homoquiral fácilmente disponible (ver sección 3.7), se exploró su posible utilidad como material de partida para la síntesis enantioespecífica de dichos segmentos. 14
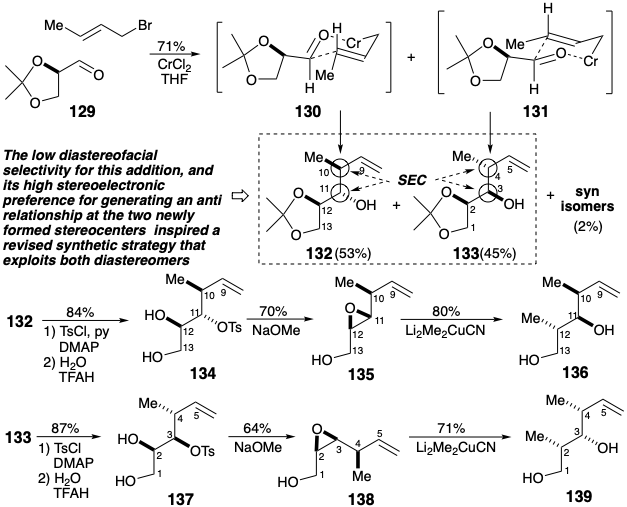
La adición de un reactivo de crotilcromo al aldehído 129 no mostró prácticamente selectividad diastereofacial para la adición al aldehído, pero una alta preferencia por generar una relación anti en los dos estereocentros recién formados debido a una preferencia estereoelectrónica por el estado de transición similar a una silla estructuras 130 y 131 que conducen a 132 y 133. Estos diastereómeros fueron fácilmente separables por cromatografía preparativa en columna a gran escala. La conversión de 132 a un intermedio de tipo 127 y de 133 a un inter- mediado de tipo 128 requiere inversión del hidroxilo secundario libre y sustitución del otro sustituyente de oxígeno secundario por metilo con inversión de configuración. La inversión del hidroxilo libre se logró mediante activación como tosilato seguido de desplazamiento intramolecular de S N 2 por un hidroxilo vecinal. Esto produjo estereoespecíficamente 135 de 134 y 138 de 137. La reacción de estos epóxidos\(\ce{Li2Me2CuCN}\) logró la segunda inversión configuracional durante el reemplazo de un sustituyente de oxígeno por metilo. Los dioles 136 y 139 corresponden a los fragmentos 127 y 128 respectivamente, donde FG 1 y FG 3 son ambos aldehídos latentes mientras que FG 2 y FG 4 son ambos grupos hidroximetilo.
La disponibilidad de los bloques de construcción homoquirales 136 y 139 canalizó una segunda fase de planeación estratégica. 15 La desconexión polar de eritronólido B en los C6-C7 y C8-C9 genera dos precursores, 140 y 142, ambos con funciones carbonilo terminales. La unión polar de estos fragmentos requeriría un “sintón de dos carbonos (C6/C7) vecinalmente dianiónico” 141 con un grupo metilo colgante. Si bien se pospuso la identificación de un equivalente sintético para 141, se reconoció que “la ramificación de metilo excluyó la aplicación directa de algún derivado acetilénico”.
La desconexión polar adicional de 142 para generar un aldehído 143, que debería estar disponible a partir del bloque de construcción 139, requiere un sintón de acetil carbanión para el cual el bromuro de isopropenilmagnesio es un equivalente sintético latente. El alqueno 144 es un equivalente latente del aldehído 140. La dislocación polar adicional de 144 sugiere un nucleófilo etílico y aldehído electrófilo 145 que debería estar disponible por oxidación de 136.
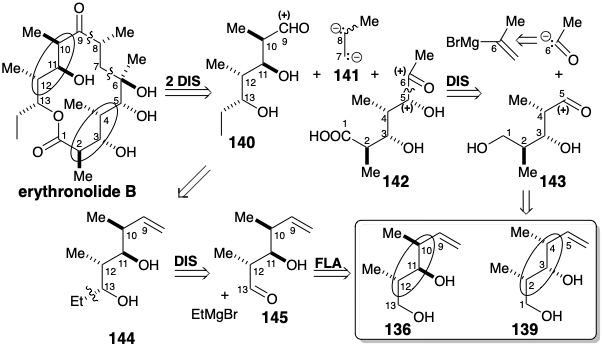
A partir del bloque de construcción homoquiral 139 se preparó un derivado enmascarado 147 de 142, que contenía la funcionalidad ácido carboxílico en forma latente como éter benciloxi, a partir del bloque de construcción homoquiral 139 (ver más adelante). La adición de un reactivo de Grignard a un intermedio aldehído generó el estereocentro en la posición 5 de manera no estereoselectiva, conduciendo a 146 como una mezcla 2:1 de diastereómeros. Sin embargo, la configuración requerida en este carbono podría establecerse mediante el equilibrio de las cetonas epiméricas 147 y 148. La cetona ecuatorial fue favorecida sobre el epímero axial 148 por 94:6 en equilibrio.
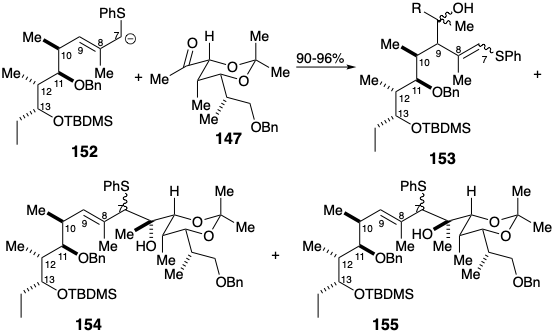
Se preparó un derivado enmascarado 149 del aldehído 140 a partir del bloque de construcción homoquiral 136. Aunque una unión polar de los dos fragmentos que contienen carbonilo 147 y 149 podría explotar un fragmento dianiónico disonante correspondiente a 141, no se ideó un equivalente sintético de 141. Más bien, un reactivo nucleófilo que era nucleófilo en C6 y contenía un carbono electrófilo en la posición 7 se unió con el aldehído 151, y luego la reactividad polar de C7 se invirtió por conversión a un tioéter alílico que podría desprotonarse para proporcionar reactividad nucleofílica en C7.

El esqueleto de carbono del eritronólido B se completó uniendo la cetona 147 con el carbanión alílico 152 producido por desprotonación del sulfuro 151 con n-BuLi en presencia de TMEDA. Los resultados iniciales fueron decepcionantes porque el producto principal fue el γ-aducto 153 más que el deseado α-aducto 154. Por adición de HMPA, la formación de 153 podría suprimirse casi por completo. Sin embargo, bajo estas condiciones, el producto principal fue un aducto α-epimérico 155. Finalmente, se descubrió que la precomplejación de la cetona 147 favoreció\(\ce{BF3}\) fuertemente la regio y estereoselecividad requeridas.
Habiendo cumplido su propósito como operador de inversión de reactividad polar, el sustituyente feniltio alílico se eliminó reductivamente. La diferenciación de los grupos hidroxilo se logró luego por acetilación seguida de desacetilación selectiva de 156 para desenmascarar el hidroxilo primario. La oxidación seguida de una desacetilación exhaustiva liberó el trihidroxiácido 157. La macrolactonización se acomodó con muy buen rendimiento por conversión a un anhídrido mixto que se cicló en solución diluida de tolueno. Aparentemente, una conformación libre de deformación que es ideal para la ciclación está disponible para 157, mientras que la congestión grave está presente en conformaciones adecuadas para formar una lactona de 12 miembros por acilación del 11-OH.
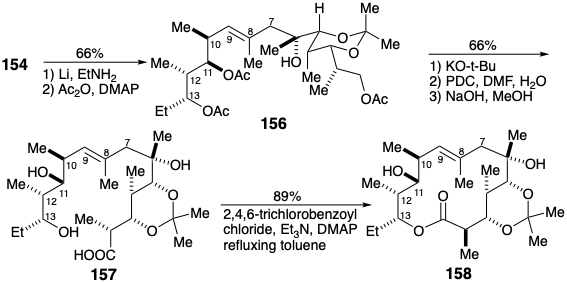
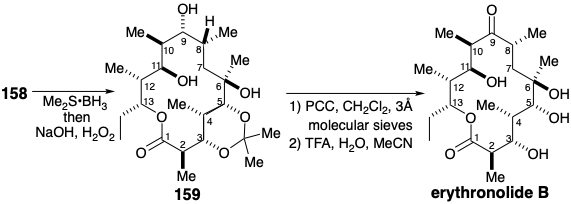
La finalización de la síntesis requirió la introducción de oxígeno en la posición 9. La hidratación anti Markovnikov del enlace 8,9-C=C en 158 por hidroboración-oxidación logró esta funcionalización, y aparentemente debido a los efectos conformacionales macrocíclicos, la generación de la configuración correcta en la posición 8 se vio favorecida por 9:1. Los efectos conformacionales también fomentaron la oxidación selectiva del hidroxilo secundario en la posición 9 en presencia de otro hidroxilo secundario en la posición 11. Así, la acumulación de muchos pasos favorablemente selectivos debido a las consecuencias sutiles e imprevistas de la forma molecular —es decir, la macrolactonización notablemente efectiva, y los procesos favorablemente estéreo y regioselectivos-, dieron como resultado una síntesis total que rivaliza con la estrategia de Corey que fue más meticulosamente planificado mediante análisis reterosintéticos exhaustivos.