6.3: Morfina
- Page ID
- 70253

Biosíntesis de Alcaloides Derivados de Bencilisoquinolina
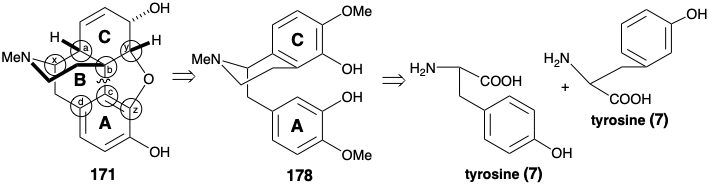
La biosíntesis de colchicina y cefalotaxina involucra a los progenitores de feniletil isoquinolina 28 y 128. Muchos alcaloides, que se derivan biológicamente de dos moléculas de tirosina, comparten progenitores de bencil isoquinolina de estructura general 169 (ver más abajo). Además, a menudo se retienen ambos anillos de seis miembrosderivados de los núcleos de arilo. Ambos pueden ser aromáticos como en berberina (170), o uno puede ser no aromático como en morfina (171). La estrategia biogenética para la berberina (170) implica una simple dislocación a un precursor de bencilisoquinolina por la desconexión de un electrófilo de un carbono del nitrógeno nucleofílico y del areno del anillo D. Una estructura de bencilisoquinolina intacta es menos evidente en el esqueleto multicíclico enrevesado de la morfina (171). Si se presume un precursor aromático rico en electrones para el anillo C altamente oxigenado, entonces la desconexión polar de un enlace furano C-O sugiere que el acoplamiento oxidativo de los anillos A y C aromáticos de un precursor de bencilisoquinolina puede generar el anillo B de 171. Los intermedios clave de bencilisoquinolina 169 podrían ser producidos por reacciones de Mannich. Estas condensaciones podrían implicar la formación de enlaces polares entre un electrófilo de fenilacetaldehído 173 y la dopamina (172) como bisnucleófilo.
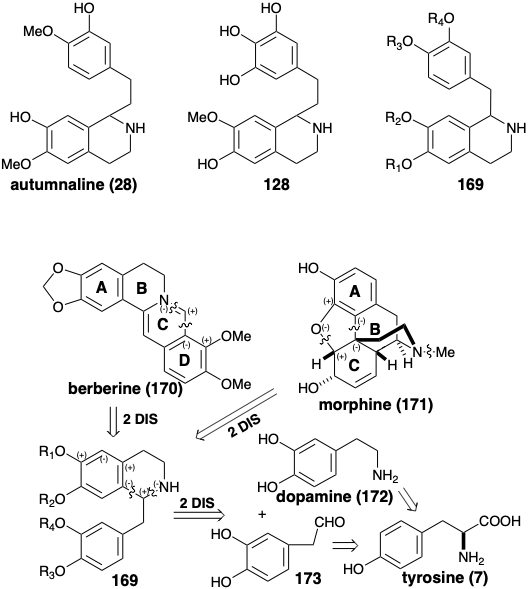
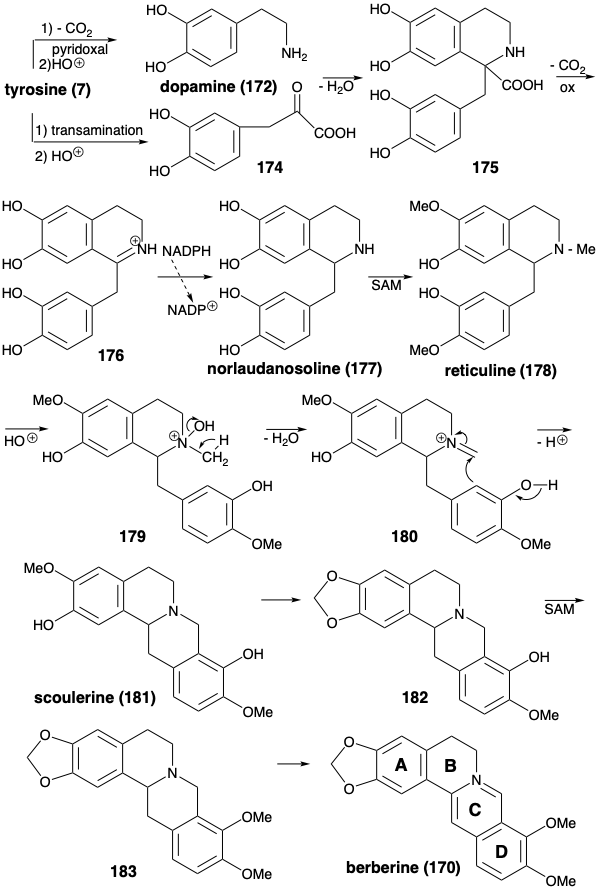
La biosíntesis de berberina (170) a partir de dos moléculas de tirosina (ver más adelante) comienza con la descarboxilación catalizada por piridoxal y la hidroxilación electrófila que produce dopamina (172). El reemplazo del grupo α-amino de la tirosina por un carbonilo por transaminación e hidroxilación electrófila produce ácido 3,4-dihidroxifenilpirúvico (174). Esta cetona altamente reactiva, más que un fenilacetaldehído, sirve como electrófilo en una reacción de Mannich con dopamina (172). La condensación polar del carbonilo altamente electrófilo en 174 con 172 como bisnucleófilo genera el sistema de anillos bencilisoquinolina en 175. La descarboxilación oxidativa de este α-aminoácido seguida de la reducción del intermedio 176 entrega norlaudanosolina (177) a partir de la cual se produce reticulina (178) por O y N-metilación. El grupo N-metilo se incorpora al esqueleto de berberina mediante una condensación de Mannich del derivado de iminio 180 producido por deshidratación de un N-óxido protonado intermedio 179. La conversión de una matriz de o-metoxifenol en el producto 181 a un grupo metilendioxi en 182, metilación y aromatización del producto 183 entrega berberina (170).
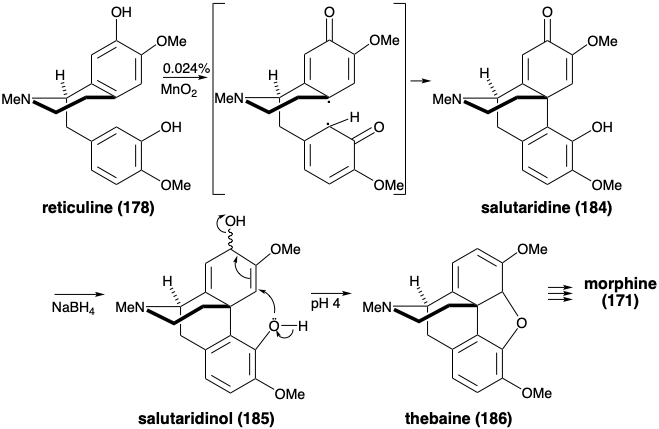
El esqueleto más topológicamente complejo de los alcaloides de la morfina también se produce a partir de la reticulina (178). Así, el acoplamiento orto-para oxidativo entrega salutaridina (184). La generación del anillo benzofurano de tebaína (186) ocurre después del ajuste del nivel de funcionalidad dando como resultado la pérdida de funcionalidad desde la posición 7 de 185. Curiosamente, aunque la hidrolisis simple del enol éter 186 podría producir cetona 188, el oxígeno del grupo metoxilo se retiene en 188. Por lo tanto, se debe involucrar un mecamismo diferente. Quizás la desmetilación de 186 ocurre a través de un intermedio oxidado 187 que sufre fragmentación retroénica. La isomerización alílica, la reducción y la desmetilación luego liberan morfina (171).
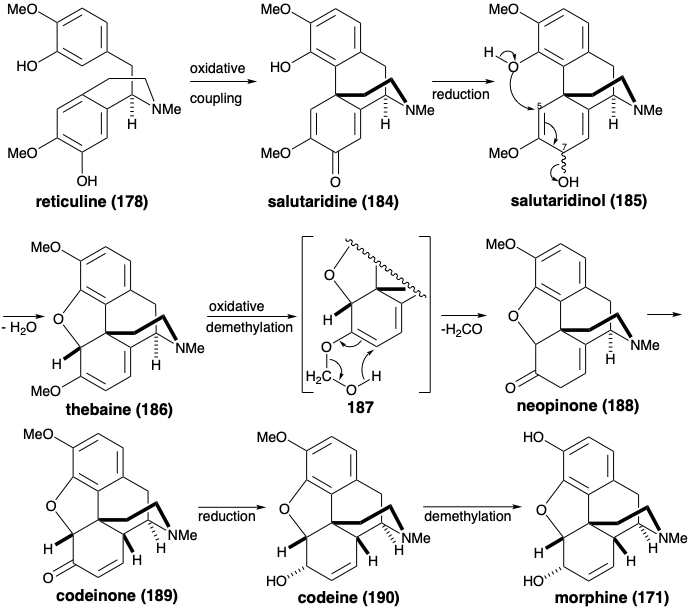
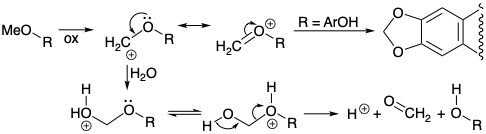
La desmetilación de codeína (190) y del éter enólico 187, así como la conversión de orto metoxi fenoles 137 en derivados metilendioxi 139 (ver sección 6.2), pueden estar relacionadas mecanísticamente por la conversión oxidativa inicial de un metilo éter en un intermedio carbocatiónico estabilizado con oxígeno-α. La desmetilación ocurriría tras la captura nucleofílica por el agua y la fragmentación del hemiacetal de formaldehído resultante.
Una síntesis biomimética de morfina
La morfina ha sido ensamblada en el laboratorio mediante una estrategia biomimética que implica acoplamiento oxidativo de reticulina (178). 9 El acoplamiento oxidativo de 178 se logró mediante tratamiento con dióxido de manganeso. Se obtuvo la salutaridina (184), aunque en rendimiento minúsculo. La reducción con hidruro proporcionó el alcohol alílico 185. Con catálisis ácida suave, 185 se sometieron a desplazamiento intramolecular S N 2' del hidroxilo alílico por el hidroxilo fenólico para proporcionar tebaína (186) a partir de la cual se puede producir morfina (171) (vide infra).
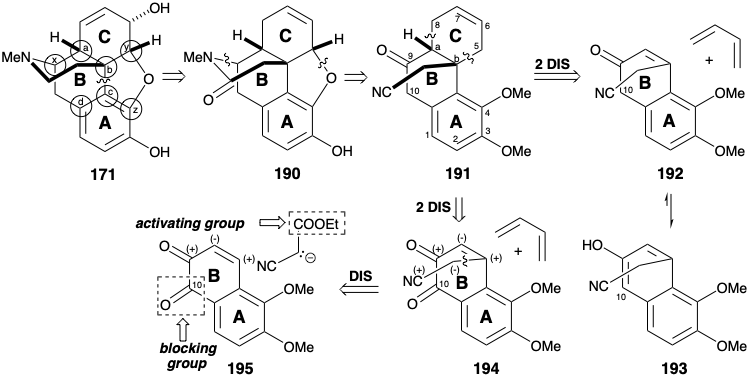
El esqueleto multicíclico puenteado de la morfina tiene una complejidad topológica considerable. Un análisis topológico (ver sección 4.4) puede, por lo tanto, ser útil para la planeación sintética. Considerando solo el esqueleto carbocíclico de 171, hay cuatro átomos comunes, a-d, y tres posibles desconexiones entre ellos. De estas desconexiones, sólo una, la eliminación del enlace entre los átomos comunes b y c, conduce a una simplificación estructural. Si también se considera el esqueleto heterocíclico, también hay tres átomos más comunes, x, y z. La desconexión de los enlaces entre estos últimos átomos comunes y un miembro de anillo heteroatómico son generalmente triviales porque los heteroátomos son funcionalidad reactiva. La desconexión del enlace b-c (y el enlace entre el átomo común y y el oxígeno) sugiere un precursor como 178 (reticulina), el progenitor biosintético de alcaloides de morfina. Curiosamente, esta es la única escisión de un enlace entre un par de átomos comunes que conduce a la simplificación del esqueleto de carbono de la morfina. Así, la escisión del enlace entre los átomos comunes a y b conduce a un intermedio con dos anillos condensados de diez miembros, lo que sería un desafío sintético indudable. Debido a que no conduce a la reducción de la complejidad molecular, esta dislocación probablemente no sea útil. La escisión del enlace π entre los átomos comunes c y d altera un sistema aromático y crea un anillo de diez miembros. La estabilidad de los sistemas aromáticos generalmente desfavorece las estrategias sintéticas que implican la anelación de anillos aromáticos en las etapas finales de una síntesis. Por lo tanto, esta dislocación también probablemente no sea útil.
Una estrategia de Diels Alder para la anulación del anillo C
La presencia de una matriz de ciclohexeno en el anillo C de la morfina (171) recomienda considerar una reacción de Diels-Alder para generar dos enlaces exendo vecinales, cada uno involucrando un átomo común (es decir, a o b) y un átomo no común. Esta simplificación topológica se aprovechó en la primera síntesis total de morfina. 10 Sin embargo, el enlace C=C en el anillo C de 171 está en la ubicación incorrecta. Por lo tanto, con el uso de una táctica Diels-Alder como condición límite, la dislocación de 171 a una amida 190 prepara el escenario para una dislocación retro de Diels Alder. Para permitir la incorporación de un enlace C=C para un dienófilo, 190 se dislocó primero a 191 por escisión de enlaces carbono-heteroátomo a los átomos comunes x e y. El carbonilo en la posición 9 en 191 proporciona activación para una construcción de Diels-Alder del anillo C a partir de un anillo AB. dienófilo 192 y 1,3-butadieno, un dieno relativamente rico en electrones. Sin embargo, esta estrategia es fatalmente defectuosa porque se espera que 192 exista casi exclusivamente en la forma 193 de enol aromático que no sería un dienófilo reactivo. Para bloquear esta indeseable enolización, se puede explotar un grupo carbonilo en la posición 10 (numeración de morfina) en un precursor 194. La cadena lateral de cianometilo se puede agregar por la unión polar de un nucleófilo estabilizado con nitrilo con el carbono β electrófilo de la matriz de carbonilo α, β-insaturado en 195. El carbonilo C-10 en 195 también facilitaría esta adición de Michael del nucleófilo de cadena lateral al evitar la enolización de la enona junto con la aromatización.
El esquema utilizado para la síntesis de 195 explota la simetría del 2,6-dihidroxinaftaleno (196) que experimenta fácilmente sustitución electrófila en la posición α. El efecto de extracción de electrones del grupo benzoilo en el monbenzoato 197 disminuye la capacidad donadora de electrones del hidroxilo benzoilado. Por lo tanto, la nitrosación ocurre regioespecíficamente en la posición α junto al hidroxilo libre. La reducción del grupo nitroso proporciona una amina 198, que se oxida a una orto quinona 199. La reducción, metilación y saponificación luego entrega fenol 200 que proporciona 195 por nitrosación regioselectiva, escisión reductora N-O y oxidación.
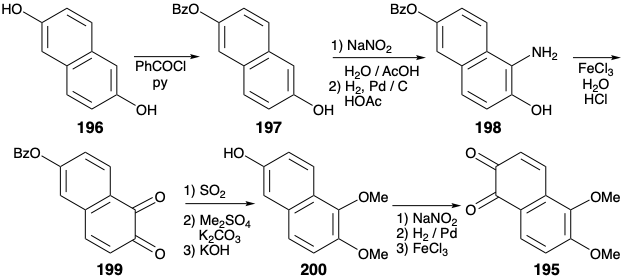
El esqueleto de carbono del dienófilo 194 se completa mediante la adición de Michael de carbanión de cianoacetato de etilo a 195. Después de la saponificación, descarboxilación y aromatización, se oxida una hidroquinona intermedia 201 para dar la orto quinona 194. El esqueleto carbocíclico de la morfina se completa con una cicloadición de Diels-Alder que proporcionó 191. La elaboración de un anillo de piperidina comenzó con una reducción que dio la lactama 202 directamente. Esta lactama es epimérica con el esqueleto de morfina en la posición 14 presumiblemente debido al control de aproximación estérica durante el suministro de hidrógeno al enlace enólico 9-14 C=C en 191.
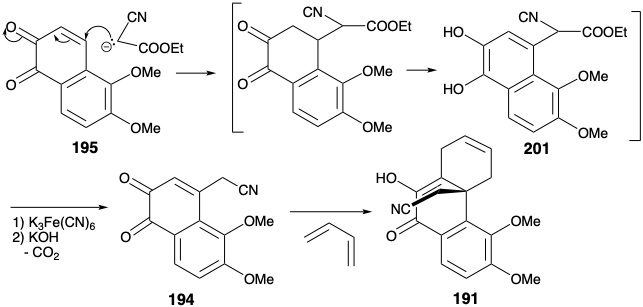
El alcohol resultante se suma presumiblemente al enlace C=N produciendo un intermedio iminoéter que se reorganiza a la lactama 202. Sorprendentemente, el enlace 9-14-C=C estéricamente mucho más congestionado se reduce mientras que el enlace 6,7-C=C permanece sin reducir bajo estas condiciones.
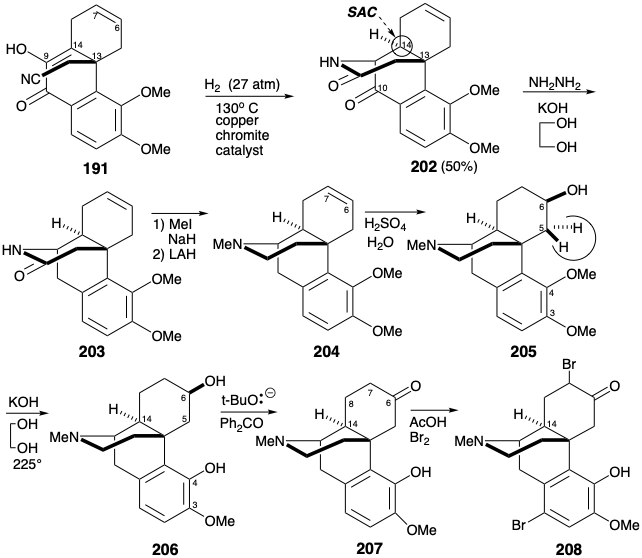
El carbonilo C-10 en 202, habiendo cumplido su propósito, fue luego eliminado por reducción de Wolff-Kishner. Después de la N-metilación de 203, la amida carbonilo se eliminó por reducción con hidruro. La hidratación del enlace π aislado en 204 procedió completamente regio y estereoselectivamente para dar 205 en “rendimientos de hasta 28%”. Esta afortunada selectividad es comprensible en términos de una preferencia estereoelectrónica por la adición diaxial antiperiplanar al enlace C=C con adición del nucleófilo preferentemente sin al sustituyente amino protonado. La intención original había sido desmetilar ambos grupos éter de 205 e intentar una remetilación selectiva del 3-hidroxilo menos congestionado estéricamente. La acción del clorhidrato de piridinio, sin embargo, no solo escindió ambos grupos éter fenólico sino que también deshidrató el alcohol secundario. Afortunadamente, se observó cierta desmetilación del éter metílico C-4 durante la conversión 202 a 203. Este descubrimiento se aprovechó desarrollando condiciones que proporcionaron 206 en 54% de rendimiento al calentar con KOH en etilenglicol. Presumiblemente la liberación de congestión estérica fomenta la desmetilación del grupo 4-metoxi mediante un desplazamiento de S N 2 de fenolato por hidróxido. La finalización del esqueleto de morfina mediante la generación de un anillo de furano requirió un ajuste considerable de funcionalidad y estereoquímica en 206. Un carbonilo en la posición 6 podría explotarse tanto para permitir la activación de la posición 5 hacia el ataque nucleofílico intramolecular por el hidroxilo C-4 como para permitir la epimerización en la posición 14. Así, la oxidación del hidroxilo C-6 en 206 por una variación de la reacción de Oppenauer dio la cetona 207. Para proporcionar la conjugación con el carbonilo C-6 necesario para permitir la epimerización en C-14, se introdujo un enlace C=C entre los carbonos 7 y 8. Así, la bromación seguida de la deshidrobromación Mattox-Kendall (ver sección 5.4) proporcionó la tosilhidrazona epimerizada 198.

Después de la hidrólisis a una enona 210 y reducción a una cetona 211, la activación en la posición 5 se logró mediante bromación con dos equivalentes de bromo. No está claro por qué 207 no podría convertirse directamente en 212 sin la intermediación de 210 y 211. La monodeshidrobromación posterior de una α, α'-dibromocetona intermedia con DNPH entregó 212. Esta se sometió a ciclación en piridina para producir benzofurano 213 después de la hidrólisis de la hidrazona. Durante la conversión 207 a 208 se introdujo un grupo bromo adventicio en el anillo A. Esto se eliminó convenientemente durante la reducción del carbonilo C-6 con hidruro de litio y aluminio para dar codeína (190), el monometil éter de morfina (171). El suministro de hidruro a 213 ocurrió estereoselectivamente desde la cara convexa más accesible estéricamente. La desmetilación de 190 se logró por desplazamiento nucleofílico por cloruro de fenol del éter protonado.
Una estrategia de anunciación de anillos B Friedel-Crafts
Otra estrategia para la síntesis de morfina (171) es canalizada por la decisión de generar el anillo B por sustitución electrófila de un nucleófilo de anillo A rico en electrones. 11 Esta táctica requiere funcionalidad carbonilo temporal en la posición 10 incipiente que tendría que ser eliminada en las etapas finales, por ejemplo, por reducción de un precursor 214. La funcionalidad de oxígeno relacionada con el objetivo en la posición 5 puede explotarse para facilitar la introducción de la funcionalidad restante del anillo C e insaturación y para facilitar la generación del heterociclo de nitrógeno por alquilación de un nucleófilo de carbono en la posición 13. Esto requiere la activación del carbono incipiente 15, es decir α a la amida carbonilo en 214, con un nucleófugo. El apéndice de un amino nucleófilo a la posición 11 en un precursor 216 para 215 no se puede lograr mediante un proceso polar ya que ninguno de los grupos cetogénicos en 216 puede proporcionar activación electrófila en la posición 11. Por otro lado, se podría añadir un electrófilo de nitrógeno a un intermedio que es nucleófilo en la posición 11 debido a la activación por la cetona carbonilo vecina, por ejemplo, por nitrosación de la cetona 216. El esqueleto carbocíclico del anillo ABC de la morfina se puede ensamblar mediante sustitución aromática intramolecular de Friedel-Crafts de un precursor 217 de anillo AC rico en electrones. El análisis polar de 217 recomienda un electrrofilo de enona a, b-insaturada 218 que proporcionaría 217 mediante la adición de un nucleófilo activado por carboxi.
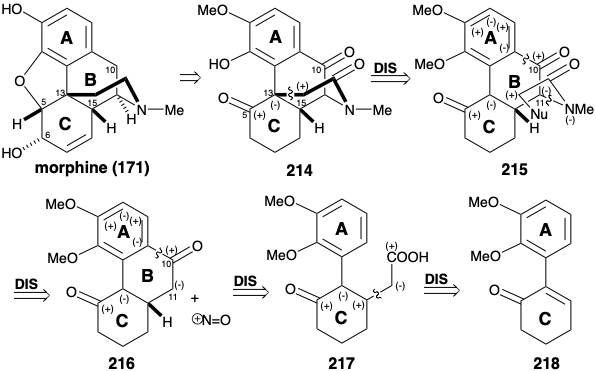
Un precursor 219 de 218 podría ser generado por subestación aromática electrófila de 220 por un electrófilo activado por carbonilo, 1,2-ciclohexanodiona. Sin embargo, la congestión estérica favorecería el producto regioisomérico alternativo 222. El uso de un nucleófilo alternativo, el diéter aromático regioselectivamente orto litiado 221 como nucleófilo, evita esta ambigüedad. Se recomiendan rutas alternativas al 218 por la posibilidad de emplear ciclohexanona como material de partida de anillo C más fácilmente disponible. Así, el arilciclohexeno 224 podría funcionalizarse mediante una secuencia de dihidroxilación-oxidación para dar 218 a 219 y 223. La posibilidad de generar 218 directamente a partir de 224 por oxidación alílica sufre de la ambigüedad de un curso regioquímico alternativo que conduce a 225.
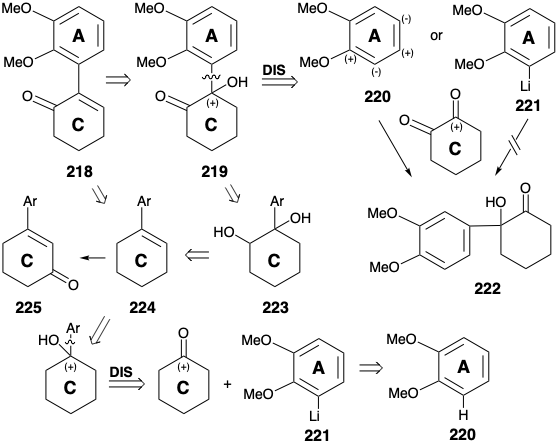
Un arilciclohexeno 224 está fácilmente disponible por orto litiación de veratrol (220) con butillitio y reacción del arillitio 221 resultante con ciclohexanona seguido de deshidratación catalizada por ácido. La bromación alílica (con NBS) o la cloración (con hipoclorito de t-butilo) seguida de hidrólisis y oxidación arrojó la enona 218 requerida. Pero este intermedio estuvo más fácilmente disponible (rendimientos globales de 40 - 50%) por adición de cloruro de nitrosilo (a partir de nitrito de amilo, ácido acético y HCl al 30%), deshidrocloración del nitrosocloruro intermedio 226 como tautómero de oxima 227 a la oxima insaturada 228 e hidrólisis.
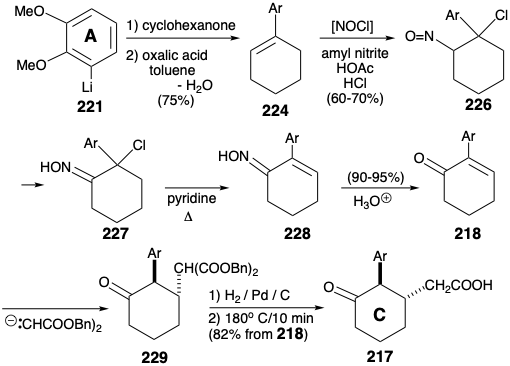
Los dos carbonos requeridos para completar el anillo B se agregaron mediante la adición de Michael de carbanión malonato de dibencilo a la enona 218, hidrogenólisis del 229 resultante y descarboxilación. La ciclación de Friedel-Crafts de 217 proporcionó el anillo B en 216. La diferenciación de los carbonilos en 216 podría lograrse mediante la cetalización selectiva del carbonilo más electrófilo. Se introdujo un sustituyente amino por nitrosación de un enolato. La reducción de la oxima 230 en condiciones ácidas estuvo acompañada de descetalización. N-acilación entregó α-acetoxiacetamida 231. La alquilación intramolecular y la cetalización selectiva (ahora del carbonilo menos congestionado estéricamente) se produjo tras el tratamiento de 231 con ácido. La transposición del carbonilo del anillo C se inició por nitrosación del enolato de 232. La descetalización seguida por la reducción de Wolff-Kishner de la dicetoxima intermedia 233 liberó la oxima 234 eliminando dos grupos carbonilo pero no el carbonilo enmascarado con oxima. La hidrólisis de la oxima, la eliminación reductora de la amida carbonilo, la metilación reductora de la amina resultante y la oxidación de un alcohol secundario intermedio liberaron la cetona 235. La conversión de un intermedio 208 análogo a morfina (171) se describió anteriormente.
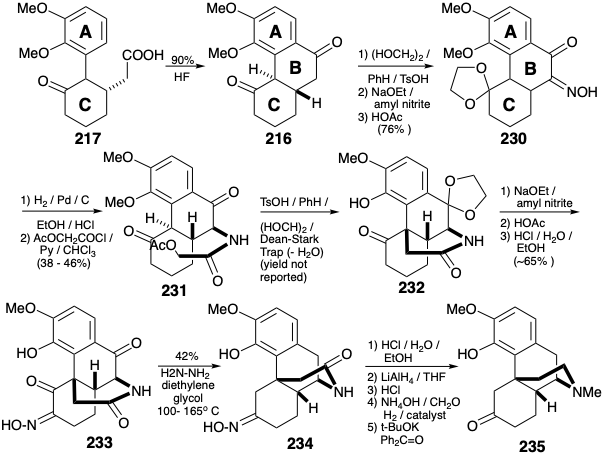
Una estrategia de adición-alquilación conjugada para la anulación del anillo B
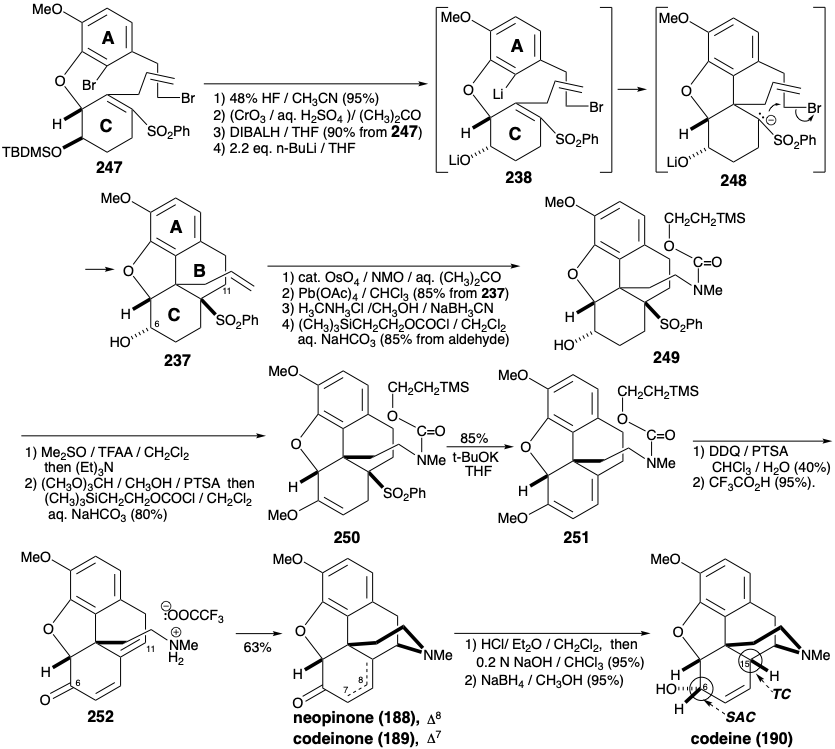
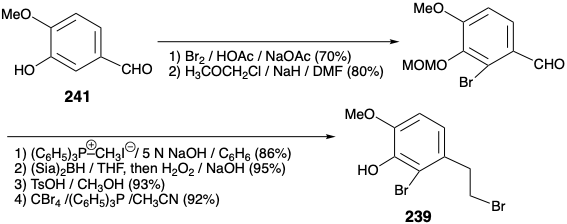
Ambas síntesis anteriores de morfina implican: (1) manipulación extensa de grupos funcionales después de la construcción de los anillos ABC y piperidina, (2) generación del último anillo furano y (3) una dependencia de grupos carbonilo para activar o controlar la reactividad. Se empleó una estrategia completamente diferente para lograr una síntesis más convergente de la morfina. 12 Esta estrategia implica: (1) generación del último anillo de piperidina, (2) manipulación mínima del grupo funcional después de completar la construcción esquelética, y (3) explotación de un grupo sulfonilo para proporcionar activación polar. En cuanto a la biosíntesis y síntesis previas de morfina, la aromaticidad del anillo A recomienda un material de partida aromático para este anillo. Un circuito consonante entre el oxígeno del anillo C y los sustituyentes de nitrógeno del anillo B en la morfina (171) sugiere una construcción del anillo de piperidina, que explota la reactividad polar proporcionada por la funcionalidad relacionada con la diana en un precursor de cetona insaturada α, β, γ, δ-236. Una doble desconexión polar del anillo B, entre un par de átomos comunes y entre un átomo común y uno no común, es posible gracias a un grupo activador de fenilsulfonilo estratégicamente colocado en un precursor 238 de 237. La desconexión polar de 238 sugiere precursores de anillo A y C 239 y 240 que deberían estar disponibles a partir de isovainillina (241) y la simétrica 2-alilciclohexano-1,3-diona (242) por adiciones e interconversiones de grupos funcionales.
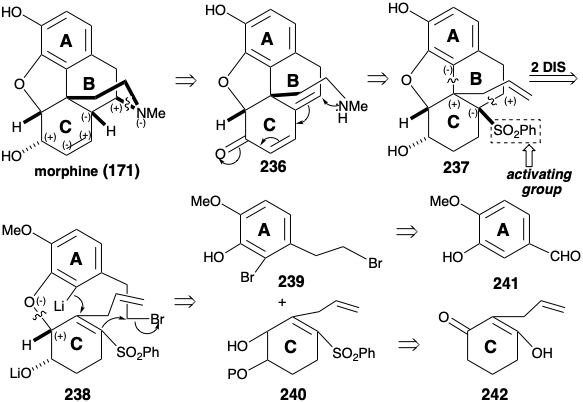
El intermedio 239 de anillo A clave está disponible a gran escala a partir de isovainillina (241) con un rendimiento total de 40% como se describe a continuación. La sustitución del hidroxilo enólico en 2-alilciclohexano-1,3-diona (242) por un grupo fenilsulfonilo se logra a través del cloruro de acilo vinilólogo 243 para proporcionar 244 mediante la adición de fenilsulfinato y cloruro de eliminación, respectivamente, en 74% de rendimiento global. La funcionalización oxidativa de 244 se realizó mediante una reacción de Rubottom, es decir, tratamiento del enol silil éter correspondiente con ácido m-cloroperbenzoico. Ni la sulfona α, β-insaturada con deficiencia de electrones ni el enlace C=C terminal se oxidan en competencia con el silil enol éter más rico en electrones. El control de aproximación estérica en una reducción de hidruro de 245 entrega el alcohol alílico 246 estereoselectivamente.
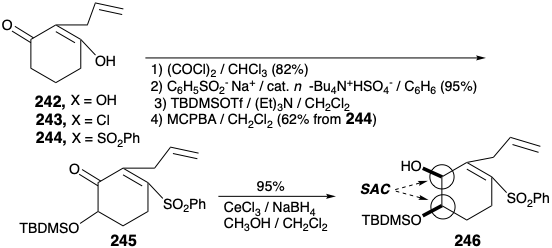
La O-alquilación de 246 con 239 proporciona el intermedio clave 247, que experimenta una notable ciclación tras el intercambio halógeno-metal. La adición intramolecular de Michael del arillitio intermedio 238 conduce a través del carbanión estabilizado con sulfona 248 a 237. La construcción del anillo de piperdina requiere la conversión del grupo alilo en 237 en una cadena lateral etilamino y la conjugación de C-11 con la funcionalidad oxígeno en la posición 6 en 237. Oxidación y eterificación enol entrega 250 de 249. La eliminación del fenilsulfinato para dar 251, y la hidrólisis para suministrar 252 establece el escenario para la finalización del sistema de anillos de morfina por generación del anillo de piperidina. Así, la neutralización de la sal de amonio 252 genera un grupo amino que se somete a adición intramolecular espontánea de 1,6-Michael a la dienona para entregar una mezcla de neopinona (188) y codeinona (189) en 63% de rendimiento. La conversión de esta mezcla a través de codeína (190) a morfina (171) se realizó como lo describió anteriormente Rapoport. La configuración requerida del hidroxilo en la posición 6 se establece durante un enfoque estérico de suministro controlado de hidruro al carbono carbonilo en 189. La configuración correcta en la posición 15 en 190 surge del equilibrio a través del derivado enol común de las cetonas 188 y 189.