6.2: Cefalotaxina
- Page ID
- 70233

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)La biosíntesis de cefalotaxina (115) implica una estrategia convergente que ensambla un intrincado esqueleto multicíclico a partir de dos precursores de aminoácidos aromáticos, fenilalanina (6) y tirosina (7). Al igual que en la biosíntesis de la colchicina (8), un anillo aromático se incorpora intacto mientras que el otro está ampliamente modificado. Así, en la biosíntesis de la colchicina (8) el anillo C de siete miembros se elabora mediante una expansión de un carbono de un anillo aromático derivado de tirosina. En contraste, la estrategia biosintética para cefalotaxina (115) explota una contracción del anillo de un carbono para producir un anillo C de cinco miembros a partir de un anillo de seis miembros derivado de fenilalanina. La lógica de la estrategia se basa en: (1) la disponibilidad inmediata de derivados ciclohexílicos altamente oxigenados como 119 por metabolismo oxidativo de precursores aromáticos y (2) la posibilidad de extruir un átomo de carbono como dióxido de carbono de una α-dicetona mediante un reordenamiento de ácido bencílico a un α- hidroxiácido. Esto sugiere el α-hidroxiácido 116 como precursor del 115.
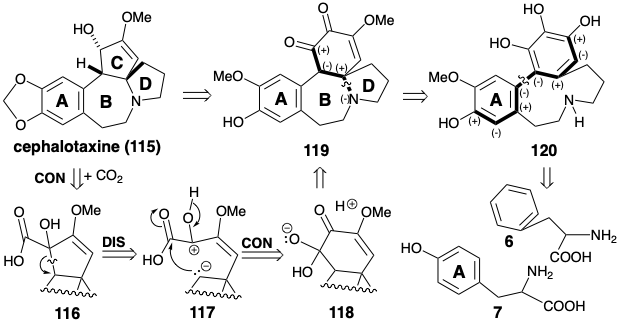
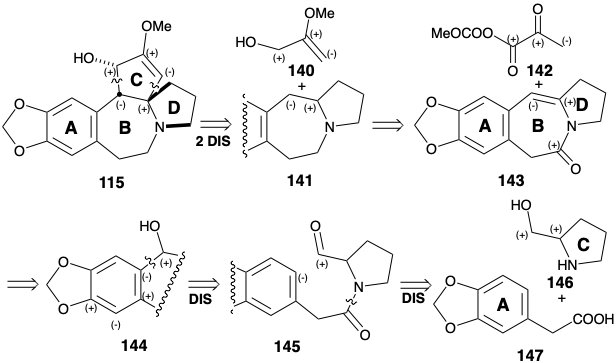
Retrosintéticamente, la dislocación de un producto 116 de reordenamiento de ácido bencílico a un precursor 119 corresponde a la desconexión polar del carbono migratorio como nucleófilo dando como resultado la oxidación del extremo de migración. La posterior conexión del carbono migratorio nucleofílico en 117 da como resultado una reducción del origen migratorio en el precursor 118. El análisis polar de 119 sugiere la desconexión polar del nitrógeno como nucleófilo de un carbono electrófilo β a un grupo carbonilo. El análisis polar del precursor 120 sugiere que los anillos aromáticos de dos precursores 6 y 7 podrían estar unidos por un acoplamiento oxidativo.
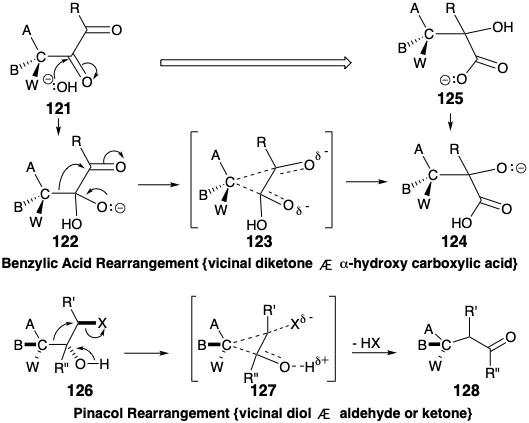
La secuencia de conexión-desconexión del reordenamiento del ácido bencílico, generalizada en la conversión 121 a 125, es mecanísticamente análoga al reordenamiento del pinacol discutido en el capítulo 4 (ver sección 3.4). El reordenamiento de 122 en 124, involucrado en el reordenamiento del ácido bencílico, es isoelectrónico con la conversión de 126 a 128 del reordenamiento del pinacol, es decir, los mismos movimientos electrónicos en matrices idénticas de átomos, enlaces, y los electrones no enlazantes están involucrados. La adición nucleofílica de hidróxido a una α-dicetona 121 inicia el reordenamiento del ácido bencílico, que procede a través de un estado de transición temporalmente puenteado 123, y finalmente produce un α-hidroxiácido 125. El reordenamiento del pinacol procede a través de un estado de transición temporalmente puenteado 127. Tanto en los reordenamientos de ácido bencílico como de pinacol, el grupo migratorio actúa como nucleófugo-nucleófilo interno que se suma a un terminal de migración electrófilo. En ambos reordenamientos el nivel de funcionalidad del origen de migración aumenta mientras que el nivel de funcionalidad del término de migración disminuye.
Se cree que la biosíntesis de cefalotaxina (115) implica el acoplamiento oxidativo de dos anillos aromáticos ricos en electrones en un intermedio 128 de fenetilisoquinolina 6 que suministra una cetona tetracíclica δ-amino-α, β- insaturada 129. La formación polar y posterior escisión polar de un heterociclo temporal de nitrógeno de seis miembros en 128, facilita el acoplamiento oxidativo al convertirlo en una ciclohexanelación intramolecular entropicamente más favorable que una ciclodecanelación que debe generar 130 directamente. Es razonable postular la presencia de un grupo metoxilo en la posición 7 en 128 ya que esto podría explicar el acoplamiento oxidativo regioselectivo en la posición 8a que es para con respecto al grupo hidroxilo que se presume está presente en la posición 6. Esta regioselectividad contrasta con la observada en el acoplamiento oxidativo del otoñalino (28) en la posición 4a (ver sección 6.1). Así, los grupos O-metilo en 28 y 128 sirven como elementos regiocontrol en los acoplamientos oxidativos de estas líneas de fenetilisoquino. La reducción y metilación regioselectiva de 130 establecieron el escenario para la activación electrofílica regioselectiva por oxidación de la orto hidroquinona 131 a una orto quinona 132.
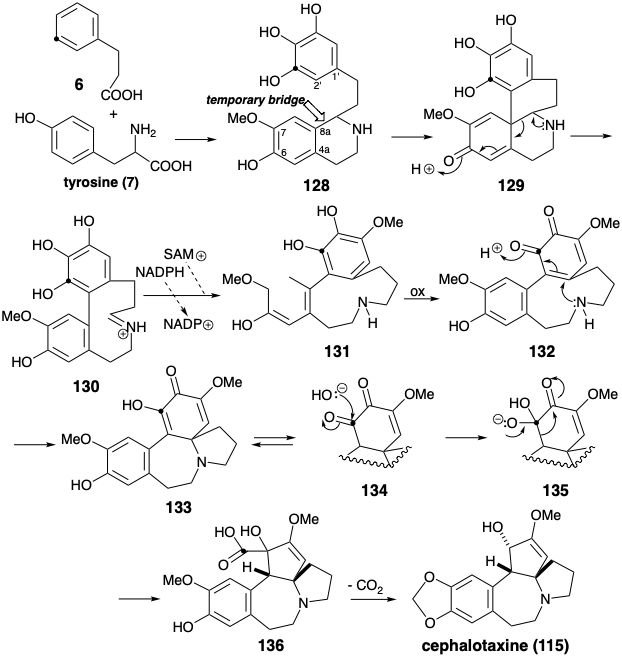
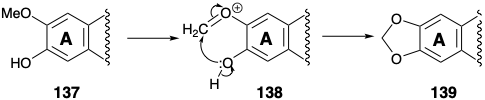
La adición nucleofílica de Michael de la amina secundaria luego entrega 133 cuyo cetotautomero 134 es una α-dicetona. El reordenamiento del ácido bencílico iniciado por la conversión a 135 proporciona α-hidroxiácido 136 en el que el carbono carboxilo se deriva de un meta carbono del material de partida de fenilalanina (6). La pérdida de este carbono como dióxido de carbono genera cefalotaxina (115) después de la conversión de la matriz orto metoxi-fenol en un grupo metilendioxi. Esta conversión, es decir 137 a 139, es común en la naturaleza y presumiblemente implica la generación oxidativa de un electrófilo 138.
Anelación del anillo B por sustitución aromática electrofílica
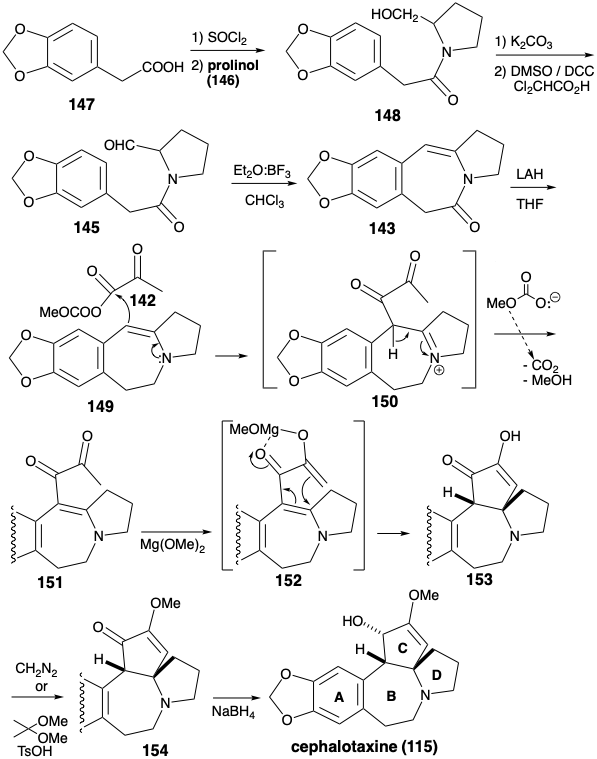
Al igual que en la biosíntesis de cefalotaxina (115), la estabilidad de derivados aromáticos recomienda la utilización de un precursor aromático para el anillo A. La estrategia de Weinreb para la construcción del esqueleto de cefalotaxina7 reconoce la utilidad potencial del grupo amino y del anillo C disonante funcionalidad para activar reacciones polares que podrían anexar el anillo C sobre un precursor de anillo ABD. Los equivalentes sintéticos 142 y 143 corresponden a los sintones polares 140 y 141. Se agrega un grupo carbonilo en 143 para facilitar la generación de un precursor 145, la amida de prolinol (146) y el ácido arilacético 147. La enamina en 143 podría producirse por deshidratación de un precursor de β-hidroxi amina 144 que, a su vez, debería estar disponible directamente por unión polar de un nucleófilo aromático y un electrófilo aldehído en 145.
La enamina 143 se construyó por anelación del anillo B entre un precursor 147 del anillo aromático A y un precursor 146 del anillo D preformado. El enmascaramiento del grupo hidroxilo en 146 es innecesario ya que la acilación ocurre en el nitrógeno más nucleófilo para dar amida 148 en lugar de en el oxígeno menos nucleófilo para producir un éster. El enlace final del anillo B se formó por sustitución aromática electrófila la cual ocurrió exclusivamente en la posición arilo menos congestionada en 145. Habiendo cumplido su propósito, la amida carbonilo se eliminó reductivamente de 143 para entregar 149. La activación polar proporcionada por el grupo acilo en 142 se explota primero para unir 142 y 149 para dar 150. Luego se explota la activación polar proporcionada por ambos grupos carbonilo para completar la anelación del anillo C. Una adición intramolecular de Michael de un anión enolato al átomo de carbono β electrófilo de un sistema carbonilo α, β-insaturado conduce a 153. La fusión de anillo cis requerida del anillo C es sin duda la más estable. El grupo lábil del anión carbonato de metilo en 142 es especialmente digno de mención. La descarboxilación de este anión genera metóxido in situ que luego desprotonata un ion iminio intermedio 150 para producir el aceptor Michael 151 en condiciones excepcionalmente suaves. También destaca el uso de metóxido de magnesio como base para generar el enolato 152 en la síntesis de cefalotaxina de Weinreb. El magnesio puede ayudar a la ciclación por quelación que impone una conformación cisoide favorable. El ajuste final de la funcionalidad implicó eterificación enólica y reducción de hidruro. El suministro de hidruro se produce desde la cara convexa menos congestionada estéricamente de 154 produciendo 115 con la configuración relativa correcta en el tercer centro asimétrico en el anillo C. Se obtuvo un éter de enol regioisomérico junto con 154. Este isómero podría reciclarse mediante equilibrio catalizado por ácido.
Anelación del anillo B por sustitución aromática nucleofílica
Mientras que la anelación del anillo B en la síntesis de cefalotaxina de Weinreb (115) se logró mediante alquilación aromática electrófila, la síntesis de Semmelhack crea la misma conexión por alquilación aromática nucleófila. 8 La estrategia de Semmelhack explota un carbonilo de anillo C para proporcionar la nucleofilia requerida en un intermedio final 155. Al igual que en la biosíntesis y la estrategia de Weinreb, se explota un precursor aromático para el anillo A. La N-alquilación de un fragmento de amina de anillo CD 157 con el fragmento de anillo A 156 proporciona 155. El mismo material de partida 147 del anillo A se utiliza para ambas síntesis totales. El anillo C en 157 contiene dos funcionalidades de oxígeno que proporcionan activación electrófila en sus respectivos átomos de carbono. Por lo tanto, estos grupos funcionales no pueden ser explotados directamente para crear el enlace entre esos átomos de carbono mediante una reacción polar. En la síntesis de Weinreb de cefalotaxina (115), el anillo C se construyó mediante reacciones polares utilizando un material de partida 142 que incorpora el circuito disonante entre las dos funcionalidades de oxígeno en el anillo C. La estrategia Semmelhack reconoce que este circuito disonante en 157 puede estar formado por una reacción no polar, acoplamiento reductivo de los dos carbonos carbonílicos electrófilos en un precursor 158. Aunque 158 podría estar disponible directamente por adición polar de dos nucleófilos de carbometoximetilo a un precursor 160 de anillo D electrófilo, Semmelhack optó por la estrategia alternativa de agregar dos nucleófilos alílicos a 160 seguido de revelación oxidativa de la grupos carboxilo latentes en un intermedio 159.
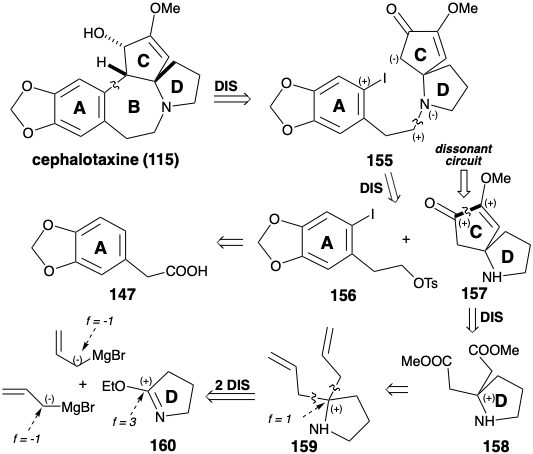
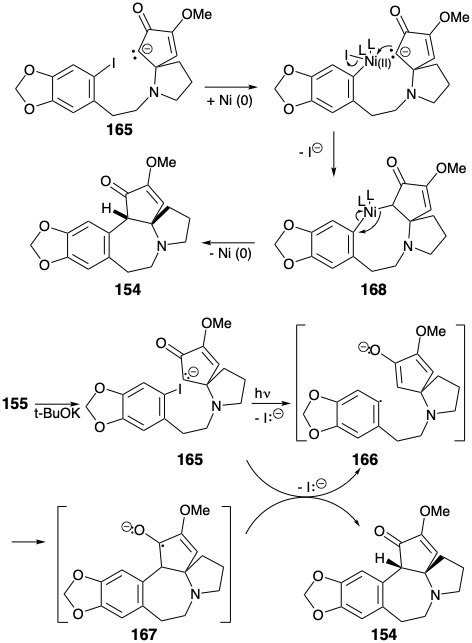
El intermedio 157 del anillo de CD se preparó a partir de pirrolidinona. La reacción de un imino éster 160 con un nucleófilo alílico dio 159. El enmascaramiento del grupo amino como una amida 161 lábil a los ácidos se requirió antes de la escisión oxidativa de los enlaces p C-C en 159 que finalmente proporcionó el diéster 158. El acoplamiento intramolecular de aciloína de 158 en presencia de clorotrimetilsilano (la modificación Rühlmann) produjo 162, que se oxidó directamente a 163 por adición de bromo en un solo recipiente y eliminación de TMSBr. La metilación de esta diona simétrica entregó 157. La alquilación de esta amina con el nitrosilato 164 proporcionó 155. Ciclización de 155, vide infra, seguida de reducción del intermedio 154 entregado cefalotaxina (115). Esta estrategia sintética conduce directamente al éter enólico correcto 154 sin formación del éter enólico regioisomérico que es un subproducto en la síntesis de Weinreb.
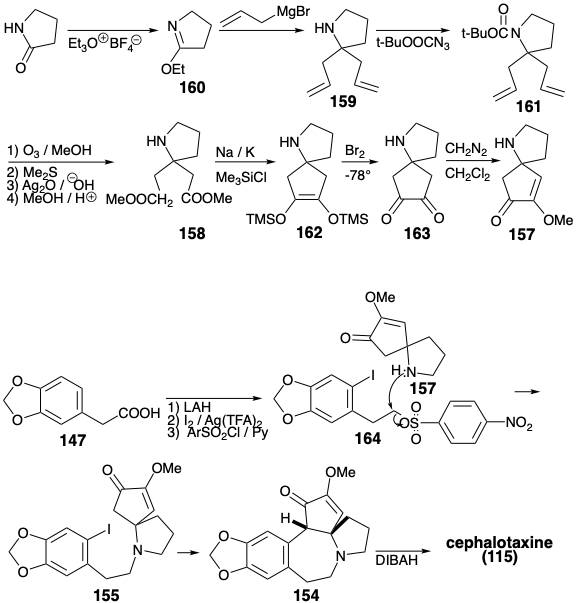
La ciclación clave de 155 a 154 se logró mediante una variedad de reacciones que involucraron la sustitución aromática nucleofílica. Por supuesto, el ataque nucleofílico directo sobre el yoduro de arilo del anillo A rico en electrones no ocurre cuando se genera un nucleófilo enolado a partir del 155. Sin embargo, la sustitución nucleofílica neta se podría lograr por fotólisis del enolato 165 o por tratamiento de 165 con Na/K o un catalizador de níquel (0). Los mejores rendimientos del producto de ciclación 154 (94%) se obtuvieron mediante una reacción de S RN 1 fotoestimulada, presumiblemente involucrando a la cadena portadora de los radicales aniónicos 166 y 167. La reacción de S RN 1 también se pudo lograr (45%) por reacción del enolato 165 con Na/K. Una reacción catalizada por níquel (0) de 165 proporcionó 154 en 30% de rendimiento presumiblemente por adición oxidativa del haluro de arilo a Ni (O) y sustitución nucleofílica de yoduro por un carbanión que produce un intermedio de σ-aril-níquel (II) 168, que sufre eliminación reductiva de 154 para regenerar el catalizador de Ni (0).