6.4: Ácido lisérgico
- Page ID
- 70243

Biosíntesis de Triptófano y Ácido Lisérgico
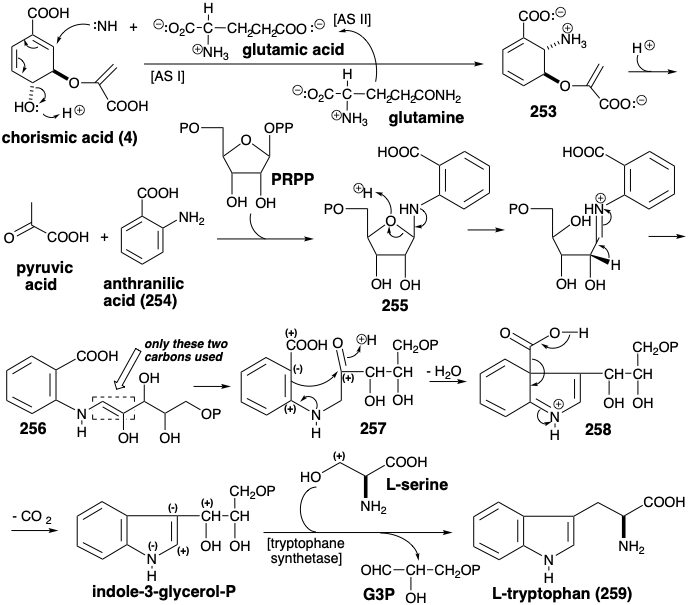
El esqueleto de carbono del ácido antranílico (254) se construye en la naturaleza a partir de dos moléculas de fosfoenol piruvato y una de eritrosa 4-fosfato vía ácido corismico (ver sección 5.1). El complejo enzimático de dos componentes de la antranilato sintetasa (AS) promueve entonces la transferencia\(\ce{NH3}\) de glutamina al ácido corismico (4) proporcionando el aminoácido 253 por desplazamiento conjugado de un grupo hidroxilo. Durante la biosíntesis estratégicamente intrincada del triptófano (259), finalmente se pierde el carbono carboxilo de 254. Una unidad de cinco carbonos de ribosa se agrega al grupo amino de 254, pero sorprendentemente los cinco carbonos del pirofosfato de fosforribosil (PRPP) no se retienen para completar el esqueleto del triptófano (vide infra). La desprotonación de la imina derivada de 255 proporciona una enamina 256 que también es el tautómero enol de la cetona 257. La ciclación intramolecular de Friedel-Crafts de este último entrega un ácido β-iminocarboxílico 258 que, siendo el análogo de nitrógeno de un β-ceto-ácido, descarboxilata fácilmente produciendo indol-3-glicerol fosfato. Si bien la biosíntesis del triptófano podría completarse con simples ajustes de funcionalidad, Nature adopta una estrategia diferente y más intrincada. Así, la triptófano sintetasa cataliza una notable alquilación de Friedel-Crafts con L-serina acoplada a una desalquilación que escinde gliceraldehído-3-fosfato (G3P) y entrega L-triptófano (259).
Hasta el momento, hemos visto que los alcaloides complejos pueden ensamblarse en la naturaleza mediante una estrategia convergente que implica la unión de dos grandes fragmentos derivados de aminoácidos aromáticos. Los alcaloides también se pueden construir en la naturaleza mediante la conjugación de intermedios derivados de aminoácidos con materiales de partida terpenoides. Así, como se describe a continuación, el ácido lisérgico (270) se forja a partir de triptófano (259) y δ 2-isopentenilo pirofosfato. Posteriormente veremos cómo el indol 259 se une de diversas maneras con la secologanina, un terpeno, para generar una amplia gama de alcaloides derivados de triptófano estructuralmente complejos. Los carbonos de estos materiales de partida permanecen conectados en el producto, aunque se pierde el carbono carboxilo. Por lo tanto, la estrategia biosintética es simple.
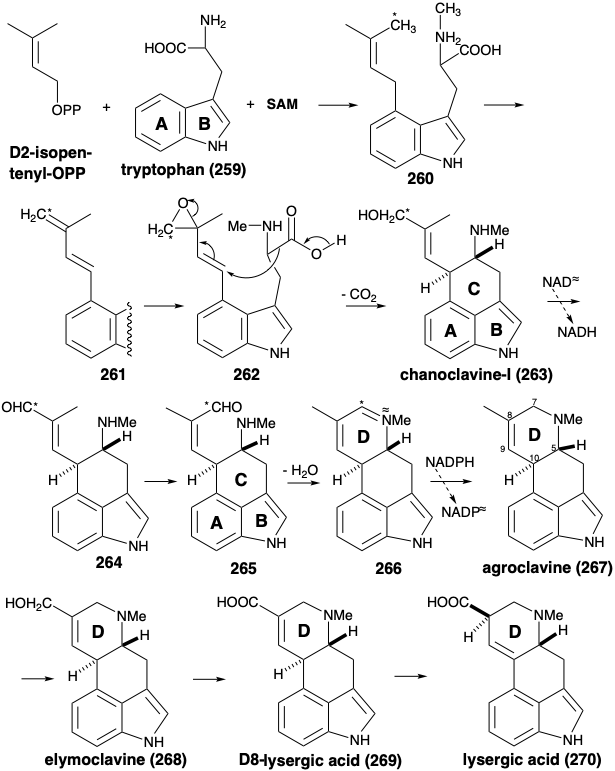
La biosíntesis del ácido lisérgico comienza con la prenilación del triptófano. Así, δ 2-isopentenilo pirofosfato es un potente electrófilo que alquila fácilmente el anillo de benceno nucleófilo del triptófano 259 para proporcionar 4-preniltriptófano (260). La ciclación de 260 a ácido lisérgico requiere la adición de funcionalidad al grupo Δ2-isopentenilo (prenilo) por oxidaciones. El proceso va acompañado de una notable odisea del carbono alílico marcado con un asterisco en los intermedios 260 - 265. La hidroxilación y deshidratación alílicas proporcionan 261. El dieno 261 tiene rotación libre, lo que permite la interconversión de los carbonos E y Z-metilo durante la conversión 260 a 263. Se cree que la formación del anillo D ocurre por una alquilación descarboxilativa de S N 2' en el epóxido alílico 262. La oxidación del producto de ciclación, Chanoclavina-I (263), a un aldehído E alílico 264 es seguida de isomerización cis-trans al isómero Z 265. La condensación a la base de Schiff 266 completa el esqueleto del ácido lisérgico. El ajuste final de la funcionalidad por reducción a agroclavina (267), oxidación alílica a elimoclavina (268), oxidación adicional a ácido δ 8-lisérgico (269) e isomerización da ácido lisérgico (270).
Dihidrógeno como grupo de enmascaramiento para un alqueno
El ácido lisérgico es termodinámicamente inestable. Los ácidos, bases o metales nobles catalizan fácilmente el reordenamiento irreversible del ácido lisérgico (270)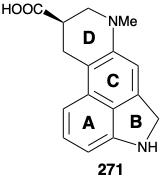 en un isómero de naftaleno 271 mediante la migración de un anillo D y un enlace C=C del anillo B en el anillo C. Por lo tanto, la estabilidad de los derivados aromáticos, que a menudo pueden explotarse ventajosamente en la síntesis molecular compleja, fue vista por Woodward como un obstáculo importante para la síntesis del ácido lisérgico (270). Una táctica central en la primera estrategia sintética exitosa, 13 fue la evitación escrupulosa de aromaticidad en el anillo C. Por otro lado, el último paso crucial del esquema, la deshidrogenación de una indolina 272, explota ingeniosamente la aromaticidad de la matriz de indol en 270 . Lo que es notable de esta estrategia es el reconocimiento de Woodward de que, aunque podría parecer poco probable que se pueda encontrar una manera de deshidrogenar 272 sin inducir también la isomerización de 270 a 271, la búsqueda de un método para lograr tal reacción selectiva podría brindar una excelente solución al reto central de la síntesis y, por lo tanto, mereció la pena el esfuerzo.
en un isómero de naftaleno 271 mediante la migración de un anillo D y un enlace C=C del anillo B en el anillo C. Por lo tanto, la estabilidad de los derivados aromáticos, que a menudo pueden explotarse ventajosamente en la síntesis molecular compleja, fue vista por Woodward como un obstáculo importante para la síntesis del ácido lisérgico (270). Una táctica central en la primera estrategia sintética exitosa, 13 fue la evitación escrupulosa de aromaticidad en el anillo C. Por otro lado, el último paso crucial del esquema, la deshidrogenación de una indolina 272, explota ingeniosamente la aromaticidad de la matriz de indol en 270 . Lo que es notable de esta estrategia es el reconocimiento de Woodward de que, aunque podría parecer poco probable que se pueda encontrar una manera de deshidrogenar 272 sin inducir también la isomerización de 270 a 271, la búsqueda de un método para lograr tal reacción selectiva podría brindar una excelente solución al reto central de la síntesis y, por lo tanto, mereció la pena el esfuerzo.
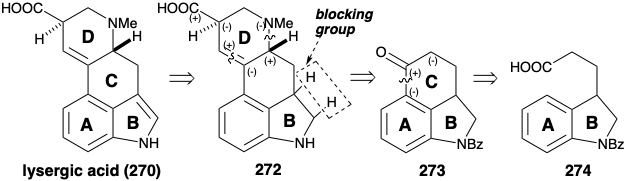
La estrategia de Woodward fue canalizada por la decisión de utilizar una acilación intramolecular Friedel-Crafts del anillo A rico en electrones en 274 para generar el anillo C en 273. Este enfoque es recomendado por la disponibilidad inmediata de 274 como material de partida, y por la utilidad potencial del grupo carbonilo en 273 para activar las reacciones de formación de enlaces requeridas para agregar el anillo D. Sin embargo, la construcción de 272 a 273 se puede lograr mediante reacciones polares solo si se emplean inversiones de reactividad polar, debido a que los patrones de reactividad polar de 272 y 273 son opuestos.
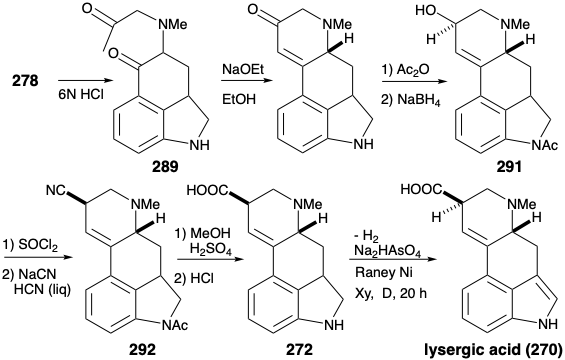
El intermedio 273 del anillo ABC está fácilmente disponible a partir de 275 por hidrogenación catalítica selectiva seguida de acilación intramolecular de Friedel-Crafts de 274. El grupo carbonilo en 273 proporciona activación en la posición metileno adyacente que puede explotarse para la unión del átomo de nitrógeno requerido. Sin embargo, un grupo amino es un nucleófilo. Para permitir la formación de enlaces C-N polares, la reactividad nucleofílica potencial del metileno α a una cetona carbonilo debe invertirse. Esto se logró por bromación para proporcionar 276 en excelente rendimiento. Un intento temprano de alquilación de la amina 277 con 276 no tuvo éxito. Después de explorar un gran número de enfoques alternativos para la anelación del anillo D y desarrollar una secuencia de once etapas para preparar 278 a partir de 273, se descubrió que un disolvente no polar era excepcionalmente efectivo para la alquilación de 277 con 276 . Bajo estas condiciones de reacción, la cetona cetal 278 se produjo con excelente rendimiento. Este escenario es un epítome conmovedor de las vicisitudes de la síntesis orgánica. Sirve para subrayar una advertencia mencionada anteriormente (ver sección 1.2) que vale la pena repetir: ya que la disponibilidad de materiales de partida o métodos (nuevos o más efectivos) para unirlos y manipularlos varía, también lo harán los méritos relativos de diferentes vías. Una mala síntesis puede convertirse en el método de elección si se descubre una forma de mejorar un mal paso.
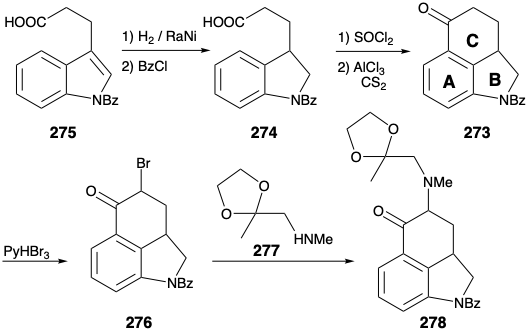
Es instructivo examinar la estrategia alternativa para la síntesis de 278 a partir del 273, y tener en cuenta que esta secuencia alternativa fue una de las muchas que fueron minuciosamente exploradas. La ruta alternativa implica una estrategia análoga a la conversión 276 → 278, excepto que el grupo carbonilo de 276 y 278 está presente en forma latente como diol vecinal en los intermedios clave correspondientes 280 y 279 respectivamente. Así, el carbonilo se genera en la última etapa de la síntesis por escisión oxidativa.
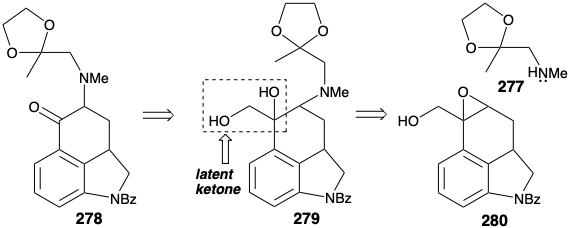
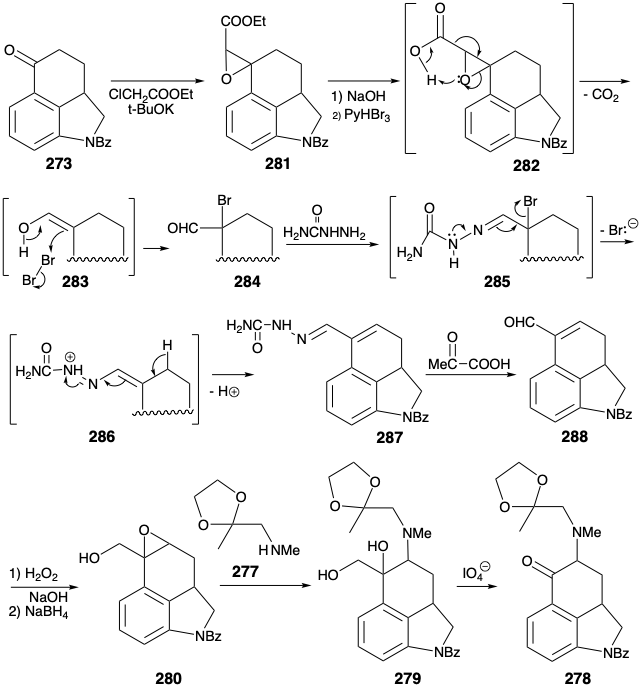
La síntesis de 279 implica una nueva secuencia en la que el enol 283 de la descarboxilación del ácido glicídico 282 es interceptado por bromo (\(\ce{Br2}\)está en equilibrio con\(\ce{Br3^-}\)) entregando α-bromoaldehído 284 del producto de condensación Darzens 281. La deshidrohalogenación del α-bromoaldehído 284 se efectuó mediante el procedimiento suave de Mattox-Kendall vía semicarbazona 285 y semicarbazona insaturada 287, que proporcionó el aldehído insaturado 288 por transferencia del residuo de semicarbazida al ácido pirúvico. La epoxidación nucleofílica entregó el intermedio clave 280. El carbonilo latente en 279 fue desbloqueado por escisión oxidativa con peryodato. Esta engorrosa ruta a 278 fue abandonada cuando se descubrieron las condiciones para lograr la preparación directa de 278 a partir de la bromocetona 276.
La terminación del anillo D fue sencilla a través de la condensación intramolecular aldólica de la dicetona 289. El grupo carbonilo en la enona 290 resultante luego proporcionó funcionalidad reactiva en un alcohol 291 y el cloruro derivado para la unión de cianuro (un equivalente de carboxi carbanión) como el átomo de carbono final del ácido lisérgico. Después de la hidrólisis de 292 a 272, los dos átomos de hidrógeno, colocados al principio por diseño en C-5 y C-5a para enmascarar un enlace C=C propenso a reordenamiento, solo necesitaron ser eliminados para proporcionar ácido lisérgico (270).
Un anillo polar D que explota la funcionalidad relacionada con el objetivo
La funcionalidad éster y amino del anillo D proporcionan patrones de reactividad polar completamente consonantes en el derivado 293 del compuesto intermedio 272 de ácido carboxílico de Woodward. Una estrategia alternativa 14 para la anelación del anillo D del ácido lisérgico explota la activación polar proporcionada por estos grupos funcionales que sugiere una dislocación a 294. La dislocación de 294 al aldehído 288, preparado previamente por Woodward, sugiere un precursor de iluro 295 en el que un éster t-butílico sirve como amina latente. Un enfoque más directo con un α-amino iluro parecía desaconsejable en vista de una posible β-eliminación.
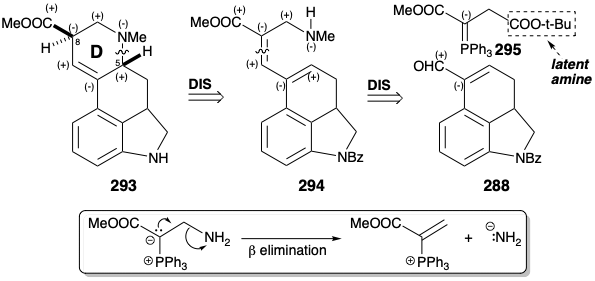
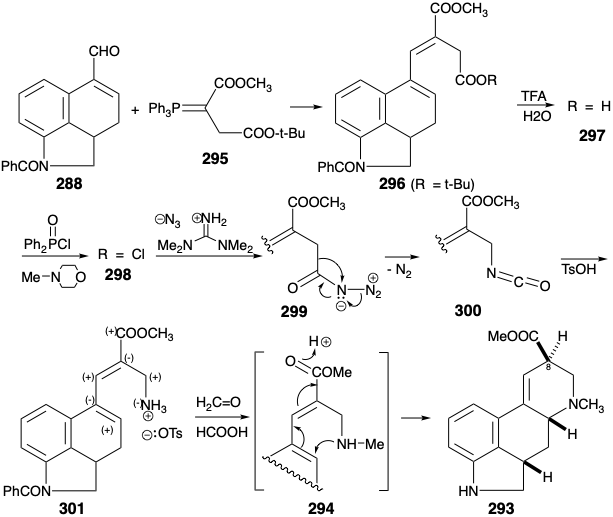
La introducción del sustituyente amino se inició por hidrólisis selectiva del éster t-butílico en 296 bajo condiciones ácidas. El ácido carboxílico 297 se transformó en la correspondiente amina primaria acortada de cadena 301 con 80% de rendimiento por una degradación de Curtius. La ciclación, es decir, de 294, acompañó la aminación reductora de formaldehído con la amina primaria 301 para proporcionar una mezcla 3:1 del éster deseado 293 y su epímero C-8.
Anulación del anillo C por sustitución aromática nucleofílica
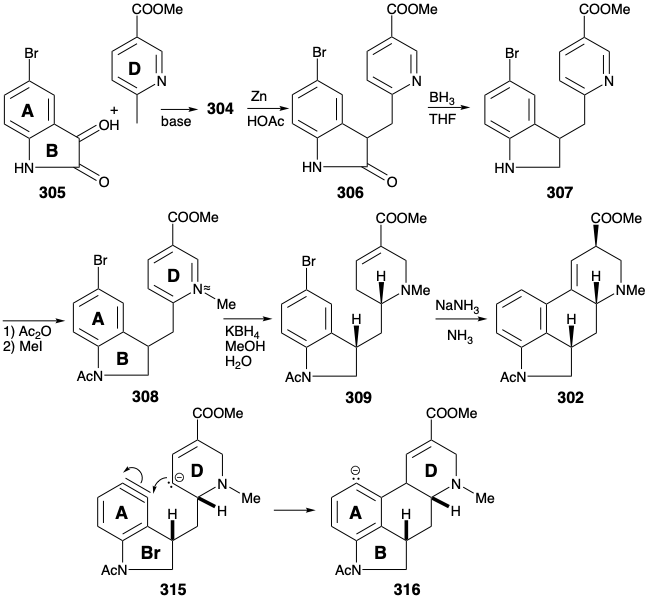
Otra síntesis del intermedio éster clave 302 implica la anulación del anillo C en un precursor de anillo ABD preformado por sustitución nucleofílica intramolecular con un carbanión 303 que se conjuga con la funcionalidad carboxilo que se encuentra en la posición 8 en la diana. 15 El mismo carboxilo también proporciona activación nucleofílica en la posición 4. Así, el análisis polar de 304 sugiere una dislocación polar a dos materiales de partida aromáticos que pueden unirse mediante una condensación aldólica entre la cetona 305 y el carbanión 306. Sin embargo, esta estrategia es fatalmente defectuosa debido a la preferencia de que la cetona 305 exista como enol. La adición de otro grupo carbonilo en la posición 2, como en 309, impide la enolización (grupo bloqueante), potencia la electrofilicidad de la cetona carbonilo, en 309, y activa el enlace C=C en 308 hacia la reducción de metal en disolución.
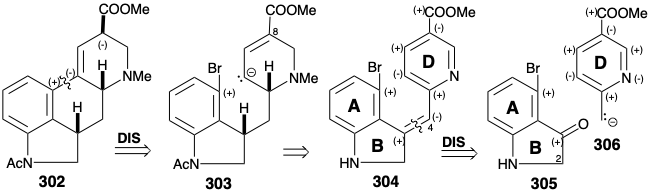
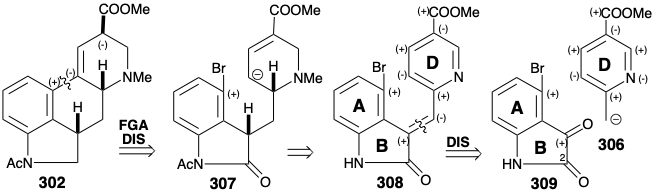
En realidad, la generación de un isómero 309, es decir 310 con el grupo bromo para a nitrógeno, se ve favorecida durante una síntesis por sustitución aromática electrófila debido a la poderosa influencia activadora del sustituyente nitrógeno. Sin embargo, esto no es un defecto porque existe un mecahanismo para la sustitución aromática nucleofílica con reordenamiento. Así, la eliminación del nucleófugo conduce a un intermedio benzino 315 al que el nucleófilo luego se agrega regioselectivamente en la posición requerida para dar 316. Habiendo cumplido sus funciones como elemento de control de la reactividad, el grupo carbonilo en 311 es luego eliminado selectivamente por reducción con diborano. El anillo de piridina del producto 312 se activa hacia la reducción de hidruro por N-metilación después de acetilación del nitrógeno del indol. Desafortunadamente, la reducción con hidruro del anillo D en 313 produce dos epímeros, solo uno de los cuales, es decir, 314, se cicla tras el tratamiento con base que suministra 302 con solo 15% de rendimiento.