6.5: Quinina
- Page ID
- 70254

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosíntesis de Alcaloides de Secologanina
Una comparación de las estructuras de alcaloides policíclicos con una variedad de esqueletos topológicamente diferentes, como la preakuammicina (317), la catarantina (318) y la tabersonina (319), sugiere una estrategia biosintética que ensambla estos heteromulticlos mediante la unión de un material de partida de aminoetil indol 400 con diez carbonos esqueléticos adicionales. El aminoetil indol, triptamina (400), se deriva razonablemente de la descarboxilación del aminoácido L-triptófano (259) mediante un proceso análogo a la descarboxilación de tirosina (7) discutido en la sección 6.4.
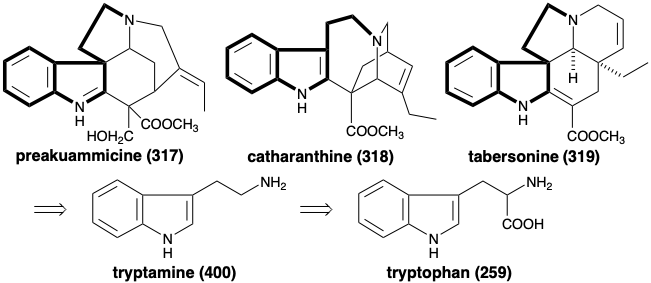
El origen de los diez carbonos esqueléticos restantes es menos obvio. El notable hecho de que estos carbonos restantes tienen un origen común está muy bien ilustrado por estudios sobre la evolución temporal de la producción de alcaloides en plántulas germinadas de Vinca Rosea. 16 Así, los alcaloides 317 - 326 y 328 son todos aislables de esta planta, mientras que 327 es un supuesto intermedio común para la generación de 318 y 319 por dos\(\pi\) cicloadiciones diferentes de 2\(\pi\) + 4.
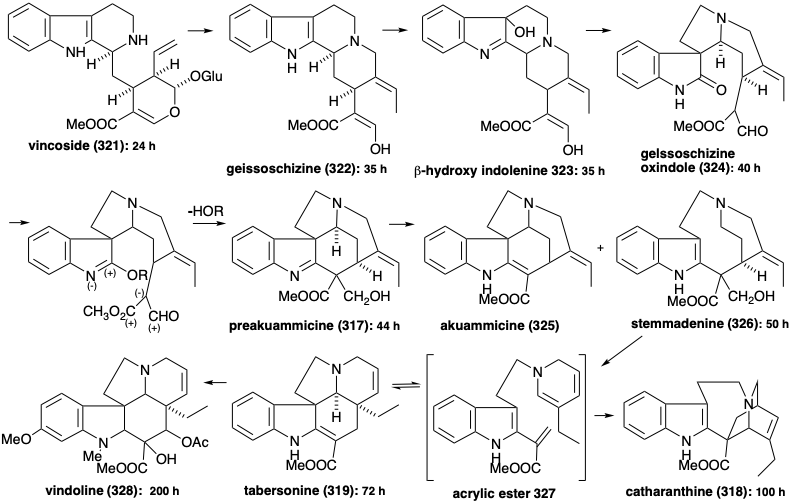
En el vincósido intermedio temprano (321), existe un circuito consonante que conecta los dos nitrógenos. Dos desconexiones polares en este circuito revelan un precursor aldehído que se parece poco a un monoterpeno además de sus diez átomos de carbono esqueléticos. Sin embargo, este aldehído es la secologanina cuyo origen terpenoide se discutió en el capítulo 4 (ver sección 4.4).
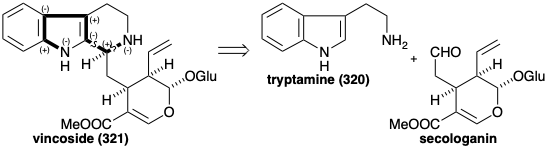
La biosíntesis de más de 1,000 alcaloides indol a partir de triptófano (259) comienza con una reacción de Mannich entre triptamina (320) y secologanina para producir vincósido (321). La hidrólisis del glucósido 321 y la descetalización del hemicetal 329 resultante proporciona el amino-aldehído 330. La ciclación de este último da un derivado de iminio 331. La adición intramolecular de Michael de un nucleófilo de oxígeno seguido de reducción proporciona otro producto aislable, la ajmalicina (332). Alternativamente, la reducción de 331 proporciona 322.
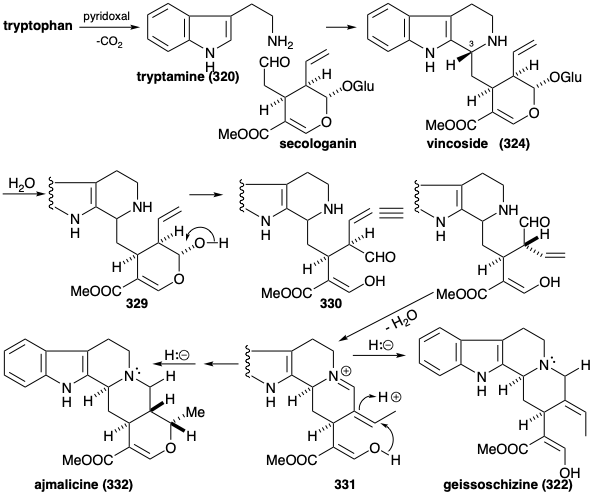
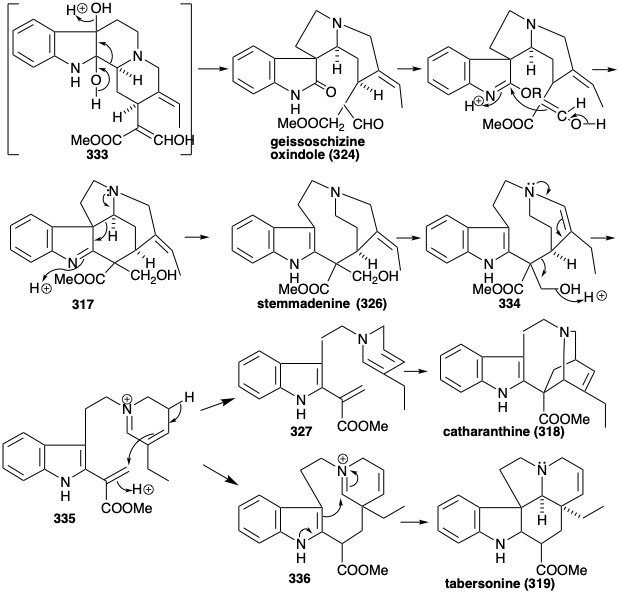
La generación de los otros tipos esqueléticos a partir de 322 implica reordenamientos que son habilitados por la introducción oxidativa de una matriz de diol vecinal para producir 333. Esta hidroxilación vecinal se logra mediante un proceso paso a paso a través de la hidratación de un intermedio aislable, la β-hidroxiindolenina 323. Un reordenamiento de pinacol de 333 produce 324. La conversión a un imino éster dotaría a 324 de la reactividad requerida para formar preakuammicina (317) por ciclación y reducción. La fragmentación similar a Retero-aldol de 317 seguida de la reducción de la immina resultante proporciona stemmadenina (326). Una segunda fragmentación del isómero enamina 334 de 326 aparentemente produce un éster acrílico intermedio 335. El tautómero dienamínico 327 de 335 proporciona el esqueleto alcaloide iboga de catarantina (318) mediante una reacción intramolecular de Diels-Alder (no necesariamente concertada). Alternativamente, el esqueleto alcaloide de aspidosperma de tabersonina (319) surge del éster acrílico 335 mediante ciclación de polieno a 336 y posterior ciclación similar a un aldol de esta última seguida de pérdida de protones para proporcionar tabersonina (319).
Biosíntesis de Quinina
Aún más críptica es la biosíntesis de varios alcaloides que contienen el sistema de anillos heterocíclicos de quinolina como la quinina (337). El sorprendente hecho de que los esqueletos de carbono de estos alcaloides también se deriven del triptófano y la secologanina ilustra aún más las longitudes a las que debe llegar la naturaleza para lograr la biosíntesis de algunos productos naturales debido a un inventario limitado de materiales de partida disponibles. Es un ejercicio instructivo para inferir la vía biosintética mediante análisis retrosintéticos. Dada la condición límite de un precursor de indol, se debe generar el anillo de piridina del sistema de anillos de quinolina en 337. Esto se podría lograr mediante la deshidrogenación de la imina producida a partir de un precursor de amino aldehído 338. La funcionalidad alcohol en 338 podría ser el residuo de la funcionalidad activadora electrófila en un precursor que estuvo involucrado en una conexión con el grupo amino nucleófilo en un anillo pirrol, como en el triptófano 339. Refiriéndose a la condición límite del triptófano como material de partida biosintético, el aldehído en el precursor 339 podría generarse por escisión hidrolítica de un derivado imina 340 de la cadena lateral de etilamina de triptamina. Se requiere una desconexión concomitante de un enlace con el grupo amino terciario en 339 para dejar espacio para la conexión. Refiriéndose a la condición límite del vincósido como material de partida, 340 podría surgir del acarboxi dialdehído 341 por descarboxilación polar y heterociclización.
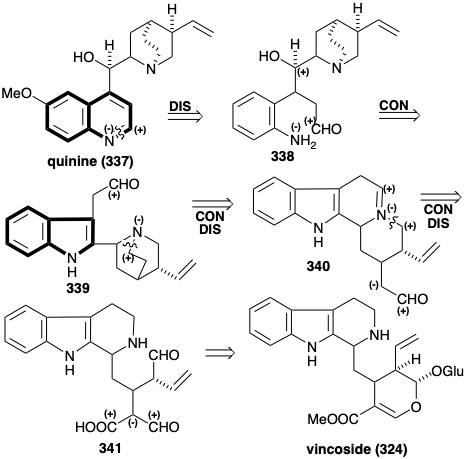
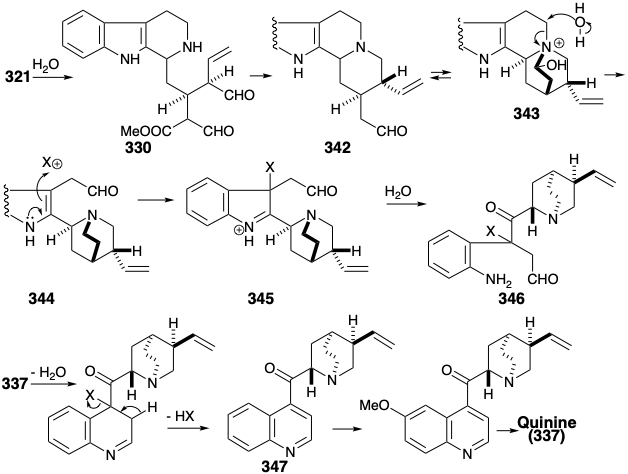
La biosíntesis de la porción quinuclidina de quinina (337) a partir de la porción de secologanina del vincósido (321) implica la hidrólisis del glucósido para dar un dialdehído 330, seguido de alquilación reductora intramolecular y decarbometioxilación para dar 342 , es decir, en equilibirum con un 343 hemiaminal. La fragmentación hidrolítica de este último acompañada por la oxidación de un alcohol primario a un aldehído y la reducción del hemiaminal a una amina da 344. El reordenamiento de la porción indol de 344 a un esqueleto de quinolina se inicia por una oxidación a 345 seguida de hidrólisis por escisión del anillo y reciclaje del amino aldehído resultante 346. Oxidación de areno, metilación y luego reducción del derivado de quinolina resultante 347 entrega quinina (337).
Una síntesis de relés de quinina
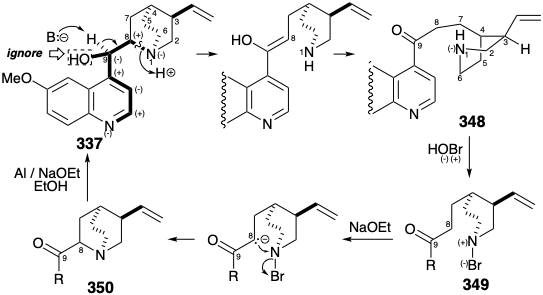
Una importante simplificación topológica del esqueleto de quinina surge por la desconexión de un enlace entre los átomos 1 y 8. El átomo 1 es un átomo común de la porción multicíclica de quinuclidina de 337. Aunque el átomo 8 es un átomo no común, su papel como vínculo entre las dos partes principales de 337 recomienda la eliminación de las conexiones esqueléticas a este átomo. Esta desconexión fue efectivamente lograda por Rabe durante estudios degradativos sobre la estructura de 337. 17 La fragmentación de 337 a 348 depende de la activación polar proporcionada por los grupos amino quinolina y quinuclidina (ignorando la activación proporcionada por el hidroxilo C-8).
Reacciones Polar Redox
Rabe también demostró que el proceso inverso, una síntesis de 331 a partir de 347, se puede lograr explotando la activación polar de C-8 en 347 (numeración quinina). 17 Este enfoque requiere un electrófilo de nitrógeno e implica oxidación a través de intermedios polares. Así, el grupo amino secundario nucleófilo en 348 se convierte en un electrófilo en 349 al añadir un átomo más electronegativo, es decir bromo. Esto constituye la oxidación del grupo amino. El ataque electrofílico al carbono en 349 para dar 350 produce un enlace entre el carbono y un átomo más electronegativo (es decir, nitrógeno). Esto constituye la oxidación del carbono acoplada a la reducción del grupo amino. Nos referiremos a tales reacciones como reacciones redox polares. Un ejemplo de este tipo de reacción ocurre en la biosíntesis de acetil CoA (ver sección 2.3) durante el ataque nucleofílico por hidroxietilideno TPP (351) sobre el disulfuro 352. Así, el carbono nucleofílico se oxida de f = 1 en 351 a f = 2 en 353 mientras que un átomo de azufre en 352 se reduce de f = -1 a f = -2 en 353. Esta reacción es compleja porque otro carbono en 351 se oxida simultáneamente de f = 2 a f = 3 en 353 junto con la reducción del segundo azufre en 352 de f = -1 a f = -2 en 353.
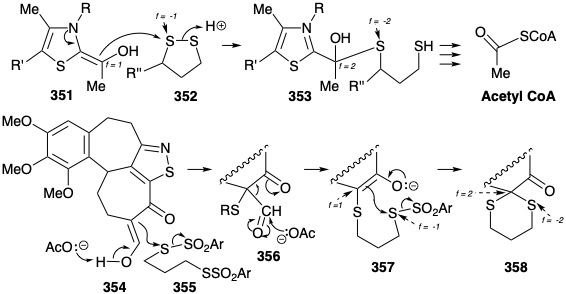
Otra reacción redox polar, nuevamente con azufre como electrófilo, se encontró en la síntesis de Woodward de colchicina (ver sección 6.1). De hecho, la introducción de un ditiocetal en el carbono nucleofílico α a un grupo cabonilo implica dos oxidaciones sucesivas del carbono α, primero de 354 a 356 luego de este último en 358, junto con dos reducciones de azufre, primero en la conversión de 355 al 356 entonces en la conversión de este último, vía 357, en 358. Esta reacción también es compleja porque otro carbono en 354, α al hidroxilo enólico, se oxida simultáneamente de f = 1 a f = 2 en 356 junto con la reducción de un segundo azufre en 355, y un segundo carbono se oxida de f = -1 en 357 a f = -2 en 358 (el carbono carbonilo) en conjunción con la reducción de un segundo azufre.
Una estrategia convergente para el intermedio clave 348
Cabe señalar que 350 (ver arriba) es una mezcla de epímeros en C-8, y la reducción que produce 337 introduce otro centro asimétrico (en C-9). Afortunadamente, 337 fue un componente principal de la mezcla isomérica producida por esta conversión no estereocontrolada de 348 a 337. Esta conversión convierte a la quinotoxina (348) en una subdiana atractiva para la síntesis total de quinina (337). La subdiana 348 se simplifica aún más por una dislocación, que rompe la molécula en dos grandes fragmentos al cortar uno de los cuatro enlaces que conectan los anillos de quinolina y piperidina. La dislocación elegida por Woodward y Doering para la primera síntesis total de quinina (337) estuvo dictada por el hecho de que el proceso inverso, síntesis de 348 de 359 y 360, tuvo un excelente precedente. Un derivado dihidro de 348 (con un grupo etilo en lugar de vinilo) fue preparado por Rabe a partir de 359 y un derivado dihidro de 360 el cual se había obtenido de la degradación de quinina natural (337).

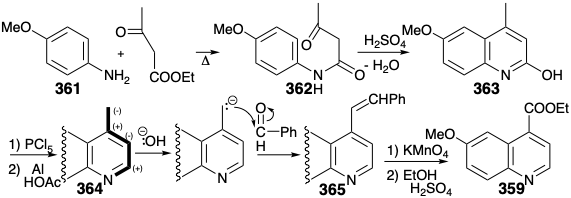
También se conocieron las síntesis totales del quininato de etilo subobjetivo (359) cuando se realizó la síntesis total de 337. Una ruta particularmente efectiva introduce el grupo carboxilo en forma latente como un grupo metilo bencílico, y construye el heterociclo de nitrógeno sobre un precursor aromático preformado 361 mediante reacciones polares. La ciclodeshidratación de 362 da 363, es decir, se reduce a 364. La oxidación bencílica de 364 se logra por escisión oxidativa de un ácido carboxílico latente, un enlace C=C en el precursor 365, que está disponible por una condensación polar de 364 con benzaldehído. La condensación explota la activación nucleofílica del metilo bencílico, que es proporcionada por el nitrógeno en 364.
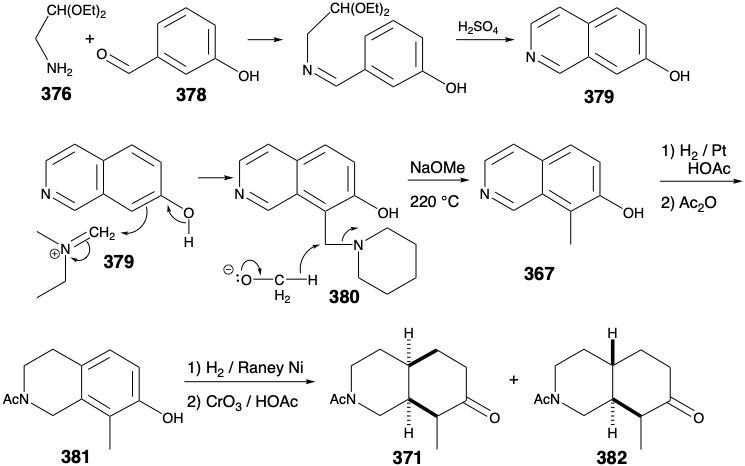
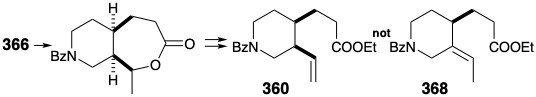
El único objetivo sintético restante fue, así, N-benzoilhomomeroquininato de etilo (360). Las dos cadenas laterales en 360 podrían generarse estereoespecíficamente cis por escisión oxidativa de un puente temporal en 366. La fusión del anillo cis en 366 podría producirse, a su vez, por hidrogenación catalítica de un precursor de isoquinolina aromática 367.

Se deben evitar varias fallas potenciales en el diseño de un esquema detallado para la conversión de 366 a 360. Por ejemplo, la escisión del puente temporal en 366 por una oxidación Baeyer-Villiger seguida de una eliminación para generar el grupo vinilo en 360 debe evitar generar el derivado alternativo, termodinámicamente favorecido, 368 de etilideno.
Curiosamente, la reacción elegida para lograr la escisión del anillo fue una reacción utilizada anteriormente en estudios de degradación para determinar la estructura de la quinina. Así, Rabe efectuó la escisión de quininona (369) en etilquininato (359) y un compuesto oximino 370 por tratamiento con nitrato de amilo y etóxido de sodio. Esta escisión es análoga a una reacción de Retro-claisen, que ocurre especialmente fácilmente para los β-ceto-ésteres no enolizables.
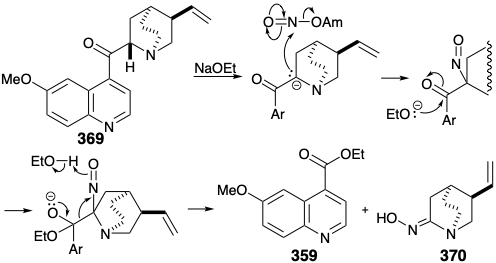
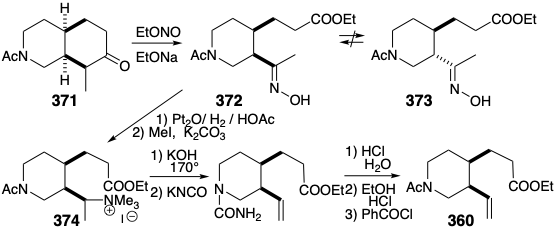
La aplicación de este proceso de escisión al análogo N-acetilo 371 de 366 genera oxima 372. Un defecto potencial, la epimerización del producto cis-1,2-disustituido 372 en el isómero trans termodinámicamente más estable 373, no fue un problema. En contraste con lo que se esperaría para el derivado acetilo correspondiente, la oxima 372 no es propensa a la epimerización. Por lo tanto, el protón hidroxilo en lugar de un protón α se abstrae preferentemente tras el tratamiento de oximas con base. La reducción y metilación de 372 proporciona fácilmente un derivado de amonio cuaternario 374, que proporciona el grupo vinilo terminal requerido en 360 mediante la eliminación de Hofmann promovida por base que implica la abstracción regioselectiva de hidrógeno del carbono β menos sustituido.
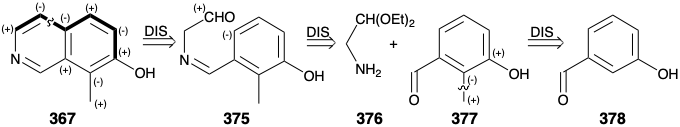
Como se señaló anteriormente, se eligió un precursor aromático 367 para la síntesis de N-benzoilhomomeroquininato de etilo (360). Un precursor aromático monocíclico para 367 podría ser un derivado de piridina o un derivado de benceno. La elección de este último permite la explotación de la sustitución aromática electrófila en un precursor rico en electrones para lograr la anulación del anillo de piridina. Esta anelación requiere enlaces carbono-carbono meta y para al grupo hidroxilo. La formación del enlace para por sustitución aromática electrófila se ve favorecida sobre meta por el fuerte efecto activador donador de electrones del grupo hidroxilo. La formación de este enlace para carbono-carbono en la última etapa de la anelación sugiere un precursor fenólico 375 con los cuatro átomos del anillo de piridina incipiente unidos a la posición meta. El enlace entre este sustituyente meta y el anillo de fenol no puede ser generado por subposición aromática electrofílica porque se favorece la sustitución orto y para en lugar de la meta sustitución. Sin embargo, la desconexión de este sustituyente por eliminación del enlace entre el nitrógeno y el carbono bencílico sugiere un aminoacetaldehído 376 enmascarado con carbonilo disonante y un derivado de benzaldehído 377. El sustituyente metilo en 377 también podría introducirse por sustitución aromática electrófila en el m-hidroxi-benzaldehído fácilmente disponible (378). Sin embargo, lograr el regiocontrol requerido en tal alquilación podría ser difícil.
De hecho, se adoptó un orden diferente de pasos. La introducción del sustituyente orto metilo se pospuso hasta que se completó la anelación del anillo de piridina, ya que la introducción de un grupo metilo se puede lograr fácilmente regioselectivamente mediante sustitución aromática electrófila en la β-hidroxi isoquinolina 379. Así, la aminometilación con piperidina y formaldehído produjo la amina bencílica 380 que se redujo a 367 al calentarse en presencia de metóxido de sodio. Esta reducción inusual implica la transferencia de hidruro del metóxido. Surgieron dificultades técnicas en la hidrogenación de 367 a 371. Así, debido a que la amina envenenó el catalizador, la hidrogenación se detuvo después de que solo se había reducido el anillo que contenía nitrógeno. La amina tuvo que bloquearse como una amida antes de que se pudiera lograr la reducción del anillo de benceno. No se pudo lograr la estereoespecifidad cis-deseada en la reducción de 381 a 371. Afortunadamente, sin embargo, esto no fue un defecto fatal porque el isómero requerido pudo aislarse del producto de reacción de hidrogenación transfusionado 383. Por lo tanto, la hidrogenación catalítica no es completamente confiable para el suministro estereoselectivo de hidrógeno a una cara de un anillo aromático.