
6.6: Biosíntesis de Aminoácidos No Aromáticos
- Page ID
- 70210

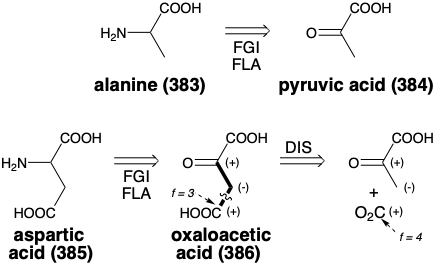 La disponibilidad de un material de partida apropiado dicta una estrategia biosintética para la alanina (383). El ácido pirúvico (384) es un intermedio clave en la biosíntesis de acetil CoA. Un intercambio de grupos funcionales (FGI) acoplado con ajuste de nivel de funcionalidad (FLA), es decir, aminación reductiva, puede proporcionar el sustituyente amino α de 383 del grupo carbonilo en el ácido pirúvico.
La disponibilidad de un material de partida apropiado dicta una estrategia biosintética para la alanina (383). El ácido pirúvico (384) es un intermedio clave en la biosíntesis de acetil CoA. Un intercambio de grupos funcionales (FGI) acoplado con ajuste de nivel de funcionalidad (FLA), es decir, aminación reductiva, puede proporcionar el sustituyente amino α de 383 del grupo carbonilo en el ácido pirúvico.
Adoptar una estrategia similar para la biosíntesis del ácido aspártico (385) sugiere el ácido oxaloacético (386) como precursor. El circuito consonante en la matriz β-ceto-ácido de 386 sugiere una síntesis polar a partir de ácido pirúvico y dióxido de carbono.
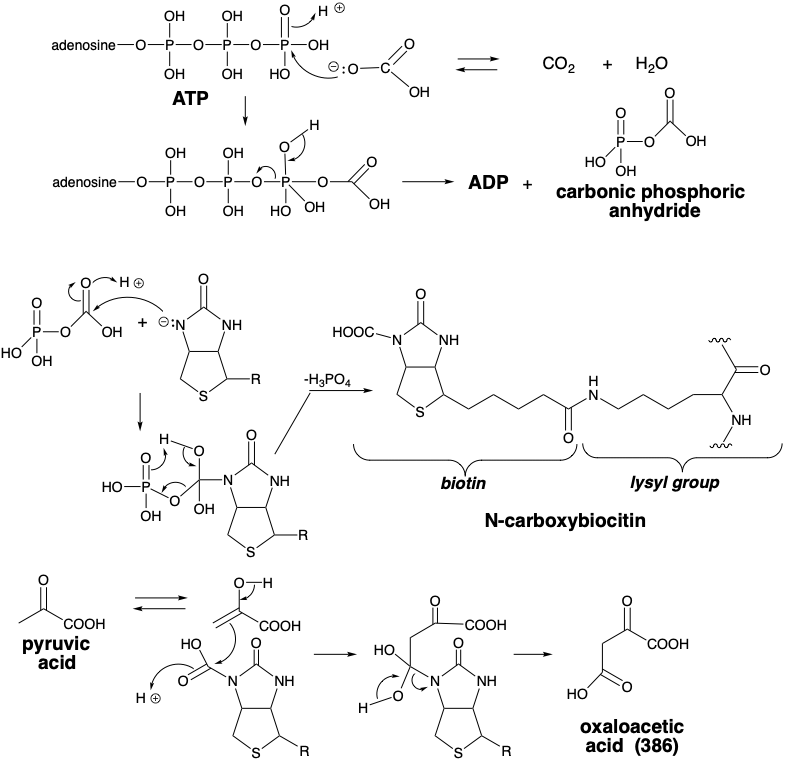
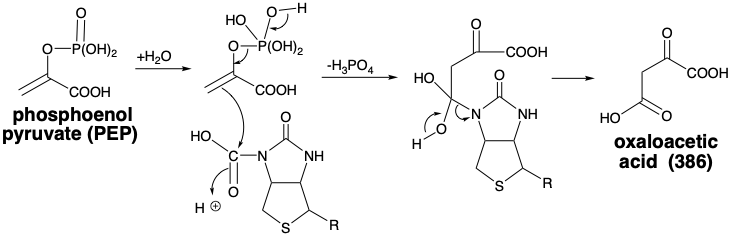
En la biosíntesis de 386, la carboxilación no implica la reacción directa del ácido pirúvico con\(\ce{CO2}\). Más bien, el agente carboxilante es un derivado de N-carboxi biotina unido a enzima que se genera por una serie de reacciones que comienzan con la activación del carbonato por fosforilación con ATP. El anhídrido fosfórico carbónico resultante acila el nitrógeno biotinílico de la N-carboxibiotina que se une a una enzima, piruvato carboxilasa, como N-carboxibiocitina. La piruvato carboxilasa cataliza la transferencia de un grupo carboxi a piruvato de N-carboxibiotina. Alternativamente, en algunas células vegetales, el piruvato de fosfoenol es carboxilado produciendo ácido oxaloacético directamente.
La estrategia biosintética para el ácido glutámico (387) es más intrincada. Así, el precursor potencial ácido α-cetoglutárico (388) contiene circuitos disonantes tanto en las matrices de ácido g como α-cetoglutárico, impidiendo una síntesis polar directa por una ruta conectiva C-C a partir de precursores más pequeños. Una forma de invertir el patrón de reactividad polar generado por un grupo funcional es 1,2-transposición de la funcionalidad. Así, el precursor transpuesto 389 tiene matrices consonantes β-hidroxiácidos que podrían ser creadas por condensación polar de un acetato C-nucleófilo con ácido malonaldehídico como electrófilo de carbonilo.
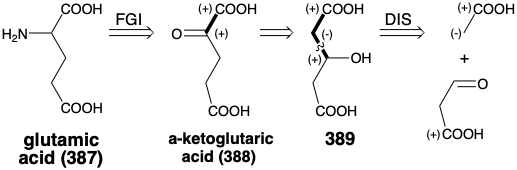
Esta estrategia polar es adoptada por Nature excepto que se utiliza como electrófilo ácido oxaloacético (386) en lugar de ácido malonaldehídico. Este material de partida tiene un grupo carboxilo extra que debe eliminarse durante la construcción del 388. La transposición 1,2-oxígeno también crea una vía polar para la descarboxilación requerida. La estrategia biosintética también tiene otras idiosincrasias. Así, además de un plan para ensamblar el esqueleto de carbono, la estrategia biosintética para el ácido glutámico incluye la cogeneración de un agente reductor. Encontramos una táctica similar en la biosíntesis de ácidos grasos a partir de glucosa (ver sección 3.1) donde la conversión de glucosa al material de partida, acetil CoA, cogenera todo el agente reductor, NADPH, requerido para la desoxigenación de los intermedios β-cetoacilo. La notable estrategia de la naturaleza para la biosíntesis de ácido glutámico genera simultáneamente un material de partida, ácido α-cetoglutárico 388, y el agente reductor requerido, NADPH, para la posterior aminación reductora de 388 para producir 387.
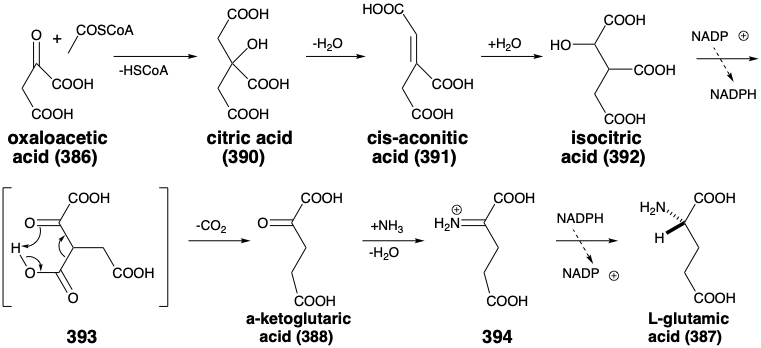
La biosíntesis comienza con la condensación de un nucleófilo acetil CoA con el carbono carbonílico altamente electofílico del ácido oxaloacético para formar ácido cítrico (390). La reacción es catalizada por la enzima citrato sintetasa (o enzima condensadora). La deshidratación del ácido cítrico da ácido cis-aconítico (391) que luego se hidrata a ácido isocitrico (392). La deshidrogenación de este último se acopla con la descarboxilación de un supuesto β-cetoácido intermedio 393 para producir ácido α-cetoglutárico (388). El hidrógeno se transfiere a NADP+ generando el NADPH necesario para la aminación reductora fijadora de nitrógeno de 388. Así, la imina protonada 394, que se produce por reacción de la cetona carbonilo con\(\ce{NH3}\), se reduce por transferencia de hidruro desde NADPH para suministrar ácido L-glutámico (387) con enantioselectividad inducida por enzimas. Este es un ejemplo de inducción asimétrica por un reactivo homoquiral (la enzima) durante la reacción de un intermedio proquiral, la imina 394.
Curiosamente, existe una vía oxidativa en la naturaleza para la conversión del ácido α-cetoglutárico (388) de nuevo en ácido oxaloacético (386). 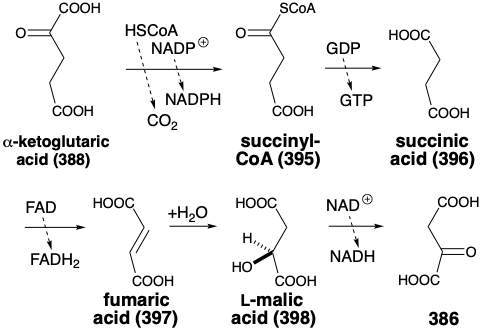 Así, la descarboxilación oxidativa del ácido α-cetoglutárico ocurre por el mismo mecanismo catalizado por pirofosfato de tiamina que para la conversión piruvato-acetato (ver sección 2.3) que cogenera NADH a partir de NAD+. El producto inicial, succinilCoA (385), es un tioéster de alta energía. Su hidrólisis se acopla con la fosforilación de ADP, a través de un proceso indirecto que implica fosforilación de un residuo de histidina de la enzima hidrolizante (ver 399), transferencia de fosfato a difosfato de guanosilo (390, GDP) y finalmente, transferencia de fosfato del GTP resultante a ADP. La deshidrogenación del ácido succínico (396) es catalizada por succinato deshidrogenasa que produce ácido fumárico (397). El hidrógeno se transfiere al dinucleótido flavina adenina (401, FAD) produciendo FADH 2. La hidratación reversible del ácido fumárico para dar ácido L-málico (398) es catalizada por fumarasa, que promueve la adición enantioselectiva estereoespecíficamente trans de agua a la olefina proquiral simétrica. Este es otro ejemplo de inducción asimétrica por un reactivo homoquiral, la enzima fumarasa. Finalmente, la L-malato deshidrogenasa cataliza la oxidación del ácido L-málico a ácido oxaloacético por transferencia de hidruro a NAD+.
Así, la descarboxilación oxidativa del ácido α-cetoglutárico ocurre por el mismo mecanismo catalizado por pirofosfato de tiamina que para la conversión piruvato-acetato (ver sección 2.3) que cogenera NADH a partir de NAD+. El producto inicial, succinilCoA (385), es un tioéster de alta energía. Su hidrólisis se acopla con la fosforilación de ADP, a través de un proceso indirecto que implica fosforilación de un residuo de histidina de la enzima hidrolizante (ver 399), transferencia de fosfato a difosfato de guanosilo (390, GDP) y finalmente, transferencia de fosfato del GTP resultante a ADP. La deshidrogenación del ácido succínico (396) es catalizada por succinato deshidrogenasa que produce ácido fumárico (397). El hidrógeno se transfiere al dinucleótido flavina adenina (401, FAD) produciendo FADH 2. La hidratación reversible del ácido fumárico para dar ácido L-málico (398) es catalizada por fumarasa, que promueve la adición enantioselectiva estereoespecíficamente trans de agua a la olefina proquiral simétrica. Este es otro ejemplo de inducción asimétrica por un reactivo homoquiral, la enzima fumarasa. Finalmente, la L-malato deshidrogenasa cataliza la oxidación del ácido L-málico a ácido oxaloacético por transferencia de hidruro a NAD+.
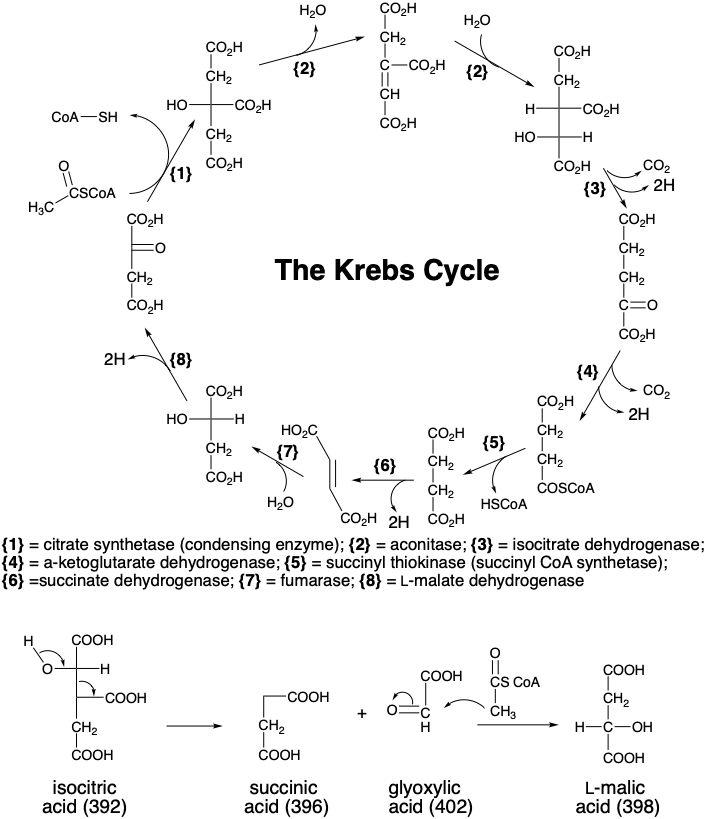
El proceso general, generación de ácido α-cetoglutárico a partir de acetil CoA más ácido oxaloacético y regeneración de ácido oxaloacético a partir de ácido α-cetoglutárico también produce dos moléculas de\(\ce{CO2}\), cuatro moléculas de agente reductor (2 x NADH, NADPH y FADH 2), y una molécula de ATP. Este ciclo de reacciones, conocido como el ciclo de Krebs o el ciclo del ácido tricarboxílico (el ácido cítrico es un ácido tricarboxílico) da como resultado el catabolismo oxidativo aeróbico del acetil CoA. Además de proporcionar una fuente de reactivos útiles para la biosíntesis a partir de ácidos grasos o azúcares (vía acetil CoA), también genera una variedad de intermedios biosintéticamente útiles, por ejemplo, ácido α-cetoglutárico) que pueden ser desviados del ciclo. Si los intermedios del ciclo de Krebs se van a eliminar del ciclo para la biosíntesis, entonces se deben generar otros intermedios del ciclo de alguna manera para reemplazarlos. La reacción anaplerótica (relleno) más importante es la carboxilación catalizada por piruvato carboxilasa de piruvato para formar oxaloacetato. Otra modificación del ciclo de Krebs, conocida como el ciclo del glioxilato, es importante en plantas y microorganismos para la producción de materiales de partida biosintéticos a partir de acetil CoA. El acetil CoA se condensa con ácido oxaloacético dando ácido isocitrico a través de ácidos cítrico y cis-aconítico. Pero, en lugar de ser oxidado a ácido α-cetoglutárico, el ácido isocitrico se escinde a ácidos succínico y glioxálico en una reacción retro-aldólica que es catalizada por la isocitrasa. El ácido succínico puede entonces ser utilizado para la biosíntesis, mientras que el ácido glioxálico (392) vuelve a entrar en el ciclo de Krebs por condensación catalizada por malato sintetasa con aceil CoA para formar ácido L-málico.
El ácido glutámico es el precursor biosintético directo de glutamina (404) y prolina (406). Activación de un carboxilo como anhídrido carboxílico fosfórico mixto (403) seguido de acilación de los\(\ce{NH3}\) suministros 404. La reducción parcial selectiva de un carboxilo y la aminación reductora intramolecular del γ-amino aldehído resultante 405 entrega 406.
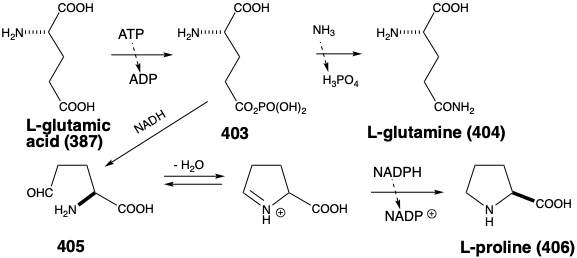
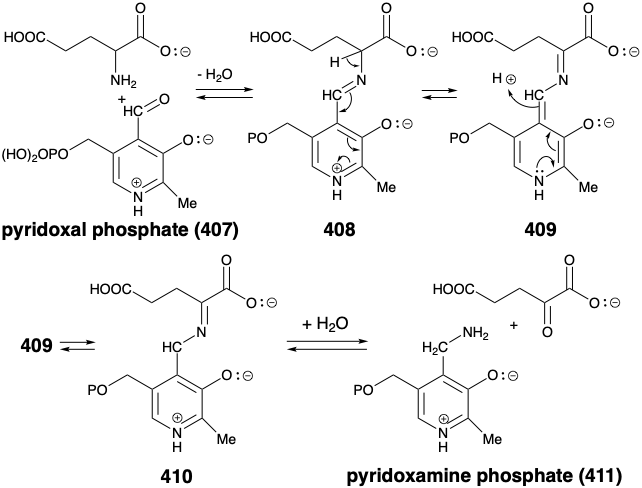
La biosíntesis de ácido glutámico a partir de ácido α-cetoglutárico por aminación reductora con amoníaco no es típica. Generalmente, la conversión de α-cetoácidos en los aminoácidos correspondientes implica la transferencia de un grupo amino del ácido glutámico, un proceso llamado transaminación. El fosfato de piridoxal (407) y los cationes metálicos divalentes son cocatalizadores para la transferencia que se produce por un proceso polar. Así, la imina de 4 0 7 y el ácido glutámico sufre desplazamiento prototrópico para generar el tautómero 4 0 9. La rearomatización por otro cambio prototrópico genera una nueva imina 410 que se hidroliza a fosfato de piridoxamina (411) y ácido α-cetoglutárico. Luego se completa la transaminación mediante la reacción de un α-ceto-ácido con 411 para producir el α-aminoácido correspondiente y regenerar piridoxal mediante un proceso que es análogo al reverso de la reacción que genera 411 más α-keto glutarato a partir de 407 más glutámico ácido.
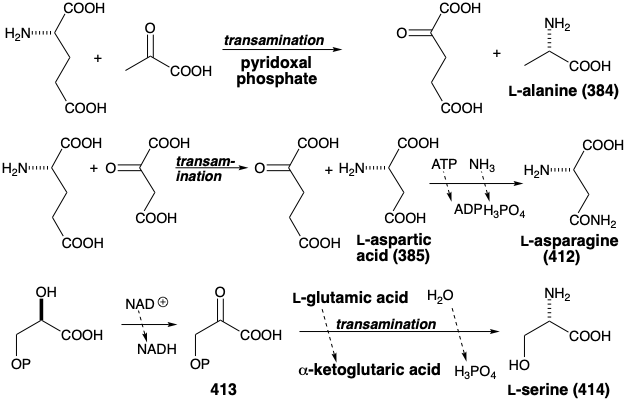
Así, la transaminación de los ácidos pirúvico y oxaloacético produce alanina (384) y ácido aspártico (385), respectivamente. La biosíntesis de asparagina (412) de 385 es paralela a la de la glutamina a partir del ácido glutámico (ver arriba). La serina (414) se produce en la naturaleza a partir de ácido 3-fosfoglicérico vía oxidación a ácido 3-fosfopirúvico (413), seguido de transaminación con ácido glutámico.
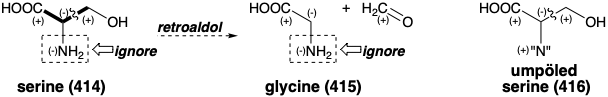
El circuito consonante entre los grupos carboxilo e hidroxilo de la serina (414) sugiere una estrategia polar para la biosíntesis de glicina (415) que implica escisión retroaldólica. El grupo α-amino está en una relación disonante con los otros grupos funcionales en 415 y, por lo tanto, no puede ayudar en la escisión polar. Sin embargo, Nature adopta una estrategia que permite que la escisión deseada ocurra en condiciones suaves por umpölung previo del grupo α-amino como en 416 que tiene dos grupos funcionales estabilizando la acumulación de densidad electrónica en el α-carbono.
El fosfato piridoxal (407) es el reactivo, un operador de inversión de reactividad polar, que convierte el grupo amino por un proceso polar en un derivado en 417 que estabiliza un anión en el α-carbono. Es instructivo considerar cómo funciona este proceso. La característica clave del reactivo 417 es una relación disonante entre el aldehído y el nitrógeno de piridinio. La reactividad electrófila del carbono aldehído se utiliza para formar un enlace C=N con el nitrógeno amino nucleófilo en serina. Luego se ignora la reactividad polar de este enlace C=N, y es el nitrógeno de piridinio disonante en 417 el que estabiliza la acumulación de densidad electrónica en el carbono serina α-carbono. El enlace C=N derivado del grupo amino de la serina sirve solo para conjugar el nitrógeno del piridinio con la serina α-carbono. La fragmentación retroaldólica de 417 produce 418 y formaldehído. Este último es capturado por ácido tetrahidrofólico (vide infra) mientras que 418 es rearomatizado y protonado para producir una imina 419 de glicina. La hidrólisis produce glicina y regenera fosfato de piridoxal que es, por lo tanto, un verdadero catalizador para la fragmentación retroaldólica.
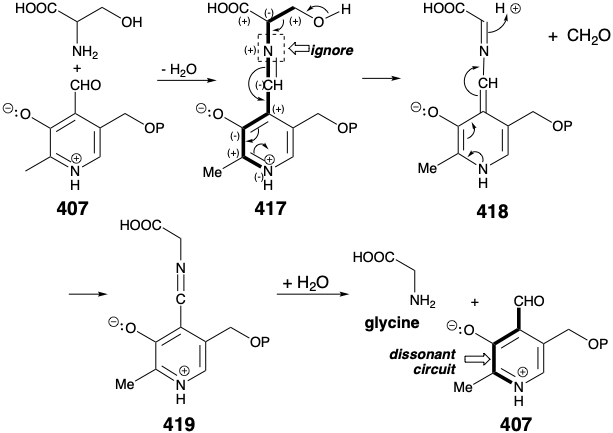
El grupo formilo perdido en la conversión de 417 a 418 se transfiere a ácido tetrahidrofólico (420, FH 4). El producto, N 5, N 10 -metileno FH 4 (421), es un miembro de una familia de coenzimas de ácido fólico que portan grupos de un carbono, como metilo, formimino y formilo en 422, 423 y 424, respectivamente.
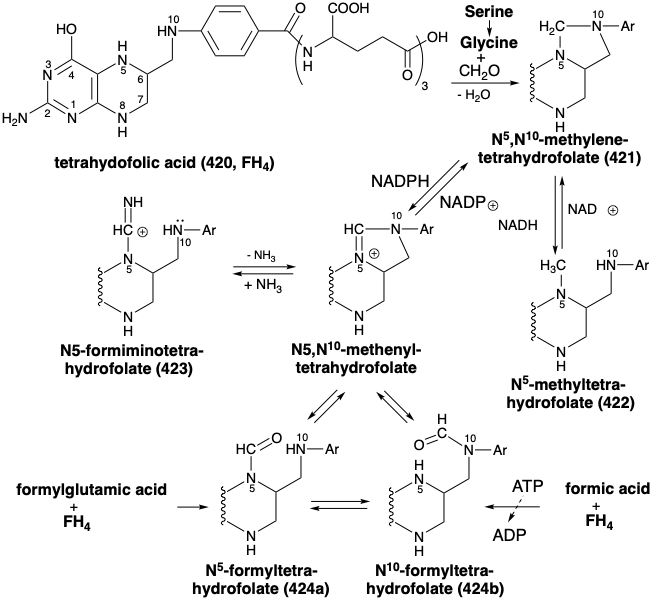
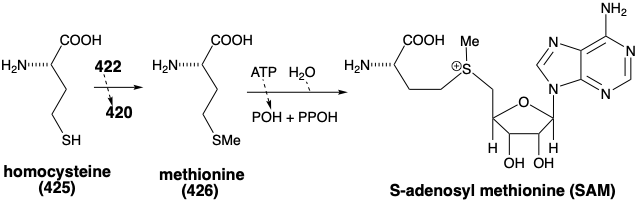
La mayor parte de estas transferencias de un carbono se logra mediante transferencia de metilo a homocisteína (425) a partir de N 5-metiltetrahidrofolato (422), lo que produce metionina (426) y, por lo tanto, S-adenosilmetionina (SAM +). Por lo tanto, la serina y, en última instancia, el ácido 3-fosfoglicérico es la fuente de los ubicuos grupos metilo donados por SAM+ a una amplia variedad de aceptores.
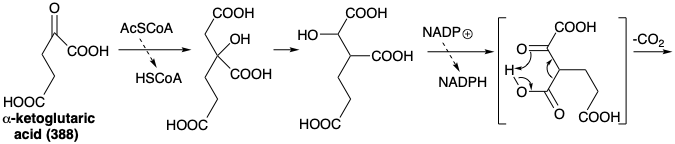
Una estrategia biosintética para la L-lisina (427) está estrechamente relacionada con la que interviene en la biosíntesis del ácido glutámico (ver arriba). Así, la doble aminación reductora de un precursor α-keto diácido 428 podría proporcionar los dos grupos amino en 427. Un precursor 429 que contiene una matriz consonante β-hidroxiácido es sugerido por 1,2-transposición de funcionalidad oxígeno en 428. El precursor 429 podría ser creado por condensación polar de un acetato C-nucleófilo con ácido succinaldehídico como electrófilo de carbonilo.
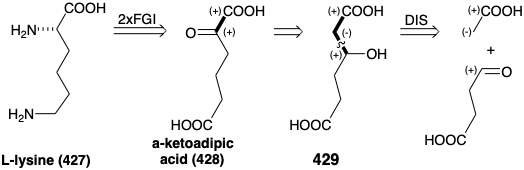
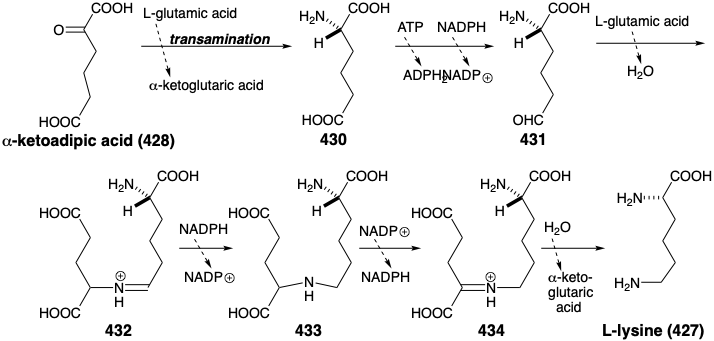
Esta es la estrategia biosintética para la lisina que es adoptada por la mayoría de los hongos excepto que se usa ácido α- cetoglutárico (388) en lugar de ácido succinaldehídico como electrófilo. La biosíntesis procede a través del ácido α-aminoadípico (430) mediante un esquema que comienza con una condensación de Claisen-Schmidt entre ácido α-cetoglutárico (388) y acetil CoA. El reordenamiento, la oxidación y la descarboxilación ocurren en reacciones análogas a la conversión del ácido oxaloacético en ácido α-cetoglutárico (ver arriba). La transaminación del ácido a-cetoadípico resultante (428) produce enantioselectivamente el isómero L del ácido α-aminoadípico (430). La reducción de 430 a 431 seguida de alquilación reductora de ácido glutámico por 431 proporciona 433. La oxidación seguida de hidrólisis da L-lisina (427). Obsérvese que el grupo iminio en 434 se estabiliza con relación al de 432 debido a la conjugación con el carboxilo en 434.
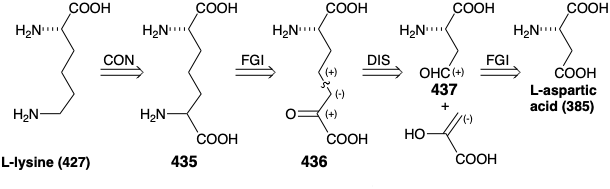
Una estrategia alternativa para la biosíntesis de L-lisina (427) genera la matriz ε-amino por descarboxilación de un grupo α-aminoácido en un precursor simétrico 435, proceso que puede ser catalizado por fosfato de piridoxal (ver abajo). Como es común en la biosíntesis de α-aminoácidos, 435 podrían derivarse de un precursor de α-ceto-ácido 436. El esqueleto de carbono de esta cetona puede ensamblarse mediante una condensación aldólica entre un enolato de ácido pirúvico y un aldehído electrófilo 437 derivado del ácido L-aspártico (385).
La vía primaria para la biosíntesis de L-lisina en bacterias y plantas superiores genera un diácido de siete carbonos intermedio 438 a partir de los tres carbonos del ácido pirúvico y cuatro carbonos del ácido aspártico (385). La reducción de 385 al aldehído 437 con hidrólisis concomitante de ATP es análoga a la reducción del ácido 3-fosfoglicérico (3PG) a gliceraldehído-3-fosfato (G3P) (ver sección 2.1) y la producción de 431 a partir de 430. La condensación aldólica de 437 con ácido pirúvico entrega 438. La formación y deshidratación intramolecular de imina produce 439 que se reducen y luego se hidrolizan y succinoilan para producir un derivado enmascarado (N-succinilado) 440 de ácido 2-amino-6-cetopimélico. La transaminación e hidrólisis de la amino amida 441 resultante proporciona la diamina 435. La monodescarboxilación de este α-aminoácido, catalizada por fosfato de piridoxal, luego entrega L-lisina (427). Al igual que en la escisión retro-aldólica catalizada por fosfato de piridoxal de serina a glicina (ver arriba), el piridoxal sirve como operador de inversión de reactividad polar que convierte temporalmente el grupo amino por un proceso polar en un derivado en 442 que estabiliza la electrónica exceso en el α-carbono. El circuito consonante entre el nitrógeno piridinio y el carbono carboxilo en 442 (ignorando la reactividad polar del grupo imina) facilita la escisión polar de un enlace C-C generando la imina 443. La aromatización por desplazamiento prototrópico produce entonces 444. Finalmente, se utiliza la reactividad polar de la imina para lograr la hidrólisis, liberando la amina 427 y regenerando el catalizador aldehído 407.
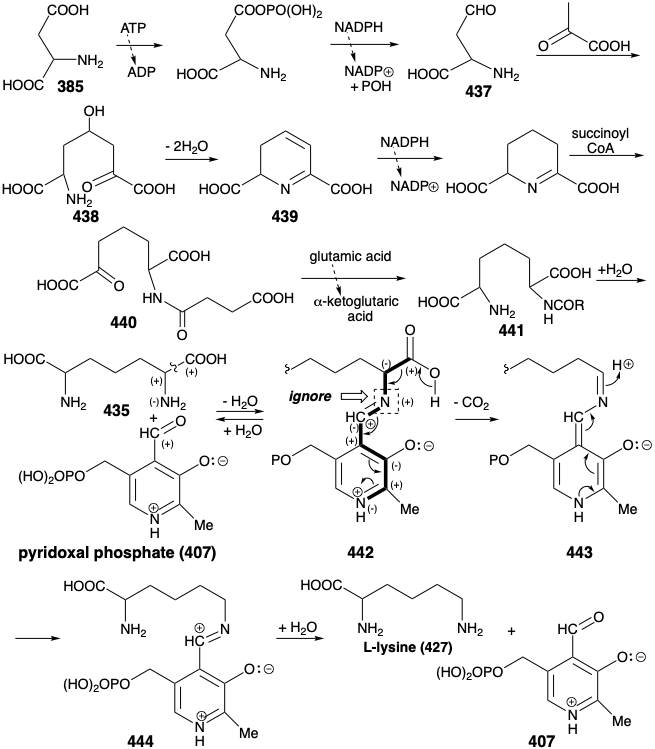
Tenga en cuenta que tanto la matriz de α-aminoácidos en 435 como la matriz de aminoácidos en 407 incorporan circuitos disonantes. Es la unión polar de un grupo funcional en cada reactivo disonante (407 y 435) lo que crea una relación consonante entre los grupos funcionales restantes en estas moléculas. Así, el umpölung del grupo amino en α-aminoácidos se logra mediante un reactivo difuncional disonante, pirofosfato de piridoxal. Anteriormente se encontró umpölung del grupo cetona carbonilo en α-cetoácidos en la condensación de benzoína catalizada por iones cianuro (sección 2.1), en la reacción de transcetolasa catalizada por pirofosfato de tiamina (TPP) (sección 2.1) y en la descarboxilación catalizada por TPP del ácido pirúvico (sección 2.2 ). Ahora es instructivo observar que tanto HC=N como TPP contienen funcionalidad bifílica. Ambos son fácilmente metalados (es decir, desprotonados). Esto introduce un grupo funcional nucleófilo (el carbanión) en una relación disonante con otra funcionalidad en estas moléculas, y les permite servir como operadores de inversión de reactividad polar catalítica. Uno de los grupos funcionales, el carbanión nucleofílico, se explota para unir el catalizador al cetol o aldehído carbonilo, en las reacciones de transcetolasa o benzoína respectivamente, o al carbonilo α-cetoácido en la reacción de descarboxilación del piruvato. La otra funcionalidad en el catalizador estabiliza entonces el exceso electrónico en el carbono anteriormente electrófilo de cetona o aldehído carbonilo.