6.7: Licopodina
- Page ID
- 70211

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosíntesis de Alcaloides de L-Lisina
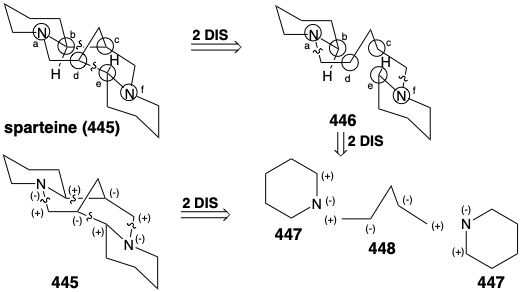
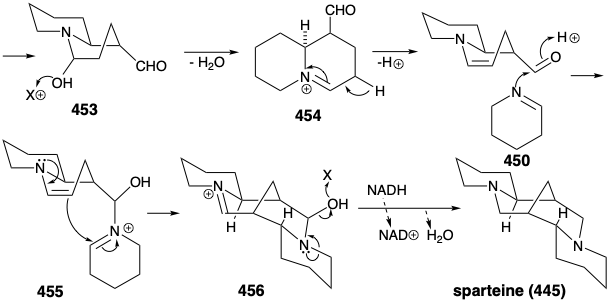
Una variedad de heterociclos de nitrógeno saturado topolgicamente complejos se construyen en la naturaleza a partir de precursores acíclicos simples. La lógica incisiva de estas biosíntesis es especialmente llamativa cuando se ve desde el punto de vista topológico o de reactividad polar. Por ejemplo, es notable la eficiencia con la que se ensambla el intrincado esqueleto multicíclico de esparteína (445), exclusivamente a partir de tres moléculas de un sintón simétrico. El análisis topológico de 445 revela la presencia de seis átomos comunes a-f. La escisión de dos enlaces entre dos pares de átomos comunes, b-c y d-e, simplifica la topología a dos anillos de piperidina unidos por una cadena lineal en 446. Este intermedio se deriva fácilmente de dos sintones de cinco carbonos 447 y 448. El análisis de reactividad polar de 445 revela que las reacciones polares, activadas por los grupos amino en 445, deberían permitir fácilmente su construcción a partir de 447 y 448.
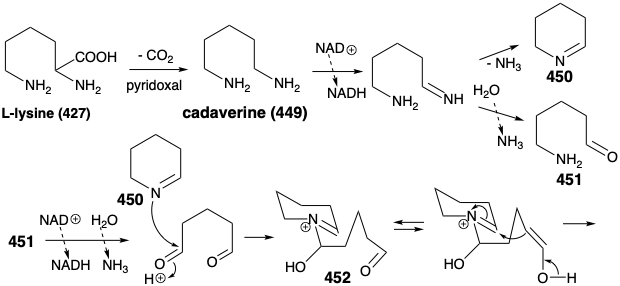
De hecho, los equivalentes sintéticos funcionalizados tanto para 447 como para 448 se preparan en la naturaleza a partir de L-lisina (427). Así, la descarboxilación catalizada por piridoxal de 427 produce la diamina simétrica 449. La oxidación e hidrólisis de 449 vía 451 proporciona pentanodial, que proporciona derivado de iminio 452 por reacción con 450. La condensación intramolecular de aldol produce 453. El derivado de iminio 454 de deshidratación de 453 produce un derivado de iminio 455 por reacción de la enamina correspondiente con un segundo equivalente de la imina 450. Una segunda condensación intramolecular de aldol proporciona 456. La deshidratación y reducción proporciona esparteína (445).
Biosíntesis de Lycopodine
Hemos visto que muchos productos naturales se forman a partir de un solo material de partida, como (a) muchos policétidos, ácidos grasos o prostaglandinas de acetil CoA, (b) muchos alcaloides del ácido shikímico, o (c) terpenos del ácido mevalónico. Sin embargo, algunos productos naturales están formados por biosíntesis mixtas a partir de combinaciones de estos materiales de partida. Así, el ácido lisérgico (ver sección 6.4) surge del ácido corismico más el pirofosfato de isopentenilo derivado del ácido mevalónico más un azúcar, D-ribosa. De igual manera, los alcaloides indol (ver sección 6.5) surgen del ácido corismico más un terpeno derivado del ácido mevalónico, secologanina, más un azúcar, D-ribosa.
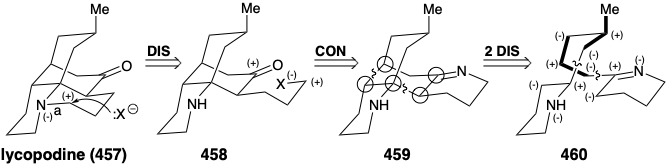
Ahora veremos que el esqueleto multicíclico puenteado del alcaloide licopodina (457) surge del acetoacetil CoA más L-lisina (427). En cuanto a la esparteína anterior, tanto el análisis topológico como el polar de la estrategia biosintética para 457 revelan su lógica incisiva. El grupo carbonilo en 457 se genera en la naturaleza por escisión solvolítica de un puente temporal. En el proceso, se genera un sustituyente propilo con activación electrófila al final, que luego se usa para construir el anillo final de 457. Retrosintéticamente, esto implica la desconexión a 458, seguida de la reconexión a 459. Una considerable simplificación de esta subdiana resulta de la desconexión de dos enlaces entre pares de átomos comunes en 459 para proporcionar 460. El análisis polar de 460 revela que la reconexión de estos enlaces podría lograrse explotando la activación polar proporcionada por los átomos de nitrógeno en 460. Además, 460 podrían ensamblarse a partir de dos fragmentos grandes mediante una reacción polar formando cualquiera de los enlaces en la cadena de carbono que conecta los dos heterociclos de nitrógeno.
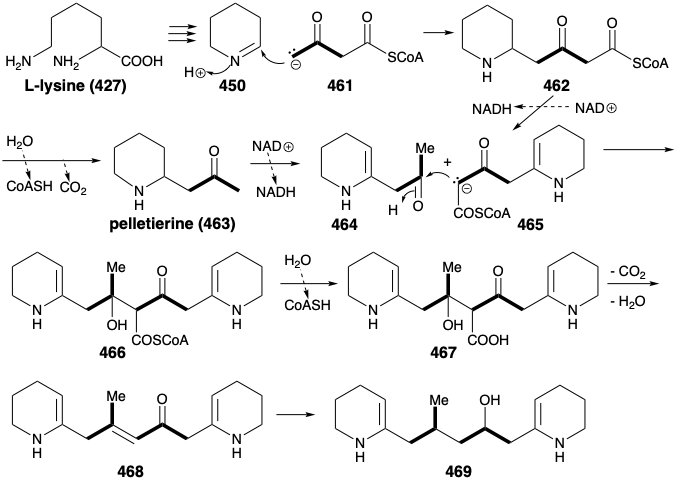
En la naturaleza, los dos anillos de piperidina en 460 se derivan de L-lisina (427), y la cadena de conexión se ensambla a partir de dos tres unidades de carbono derivadas de actetoacetil CoA (461). La condensación aldólica de 461 con 450 proporciona 462. Nótese que la alquilación de 461 ocurre en el carbono δ menos ácido. Quizás esto involucra un derivado de enamina ligado a enzima 470 (ver más abajo) de 461. La oxidación y desprotonación de 462 proporciona 465, mientras que 462 también produce pelletierina (463) por hidrólisis y descarboxilación. La condensación de aldol entre 464 y 465 luego proporciona 466, es decir, se hidroliza a 467. La eliminación descarboxilativa da 468, es decir, se reduce para proporcionar 469, un equivalente sintético del sintón 460 generado en el análisis estratégico que se presenta a continuación.
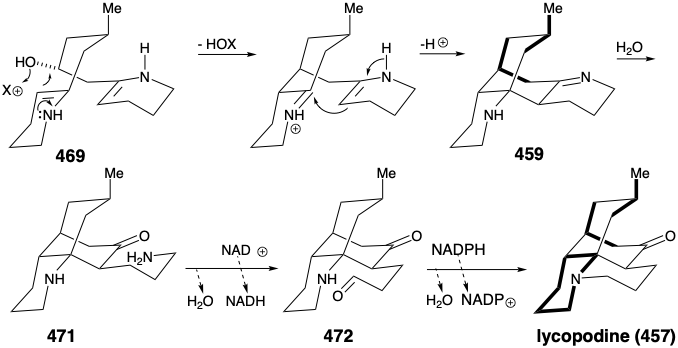
La ciclación de 469 por alquilación intramolecular enamina, seguida de condensación intramolecular similar a un aldol, produce el intermedio 459 sugerido en el análisis estratégico. La hidrólisis de la imina en 459 genera el grupo carbonilo requerido para la licopodina. La oxidación de la propilamina 471 resultante a un aldehído 472 seguida de alquilación reductora intramolecular produce entonces licopodina (457) en la que un anillo de piperidina derivado de lisina es claramente discernible mientras que las dos unidades de acetonilo derivadas de policétido y un cinco unidades de carbono de una segunda molécula de lisina están intrincadamente entretejidas.
Una estrategia fatalmente defectuosa para la síntesis de licopodina
Hemos visto que tanto el análisis topológico como el polar de la estrategia biosintética para licopodina (457) iluminan la lógica del proceso. Los dos grupos funcionales en 457 pueden ser explotados en una variedad de estrategias para facilitar la construcción de la red esquelética usando reacciones polares. Hay cinco átomos comunes en 457, cuatro átomos de carbono que están todos en el anillo B, y el nitrógeno. Primero consideraremos una estrategia fatalmente defectuosa que solo genera un epímero en lugar del producto natural en sí.
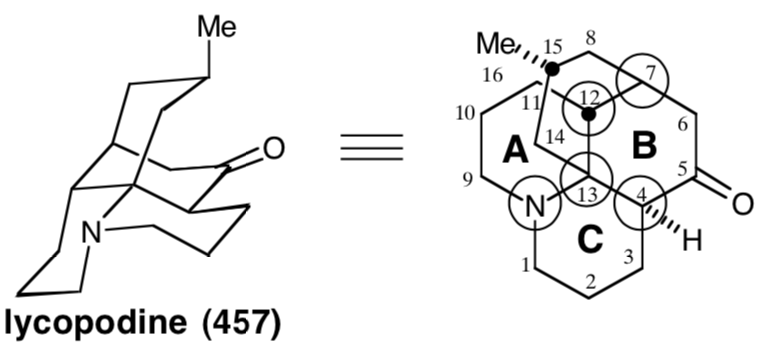
El análisis polar del anillo B revela que las reacciones polares, explotando la activación polar proporcionada por las funcionalidades amino y carbonilo en 457, podrían usarse para construir cualquier enlace de este anillo. En el enfoque Wiesner de la licopodina,19 el enlace esquelético final formado es entre el átomo común 7 y el átomo no común 6, correspondiente a la dislocación de 457 a 473. El penúltimo enlace formado es el que se encuentra entre los átomos comunes 4 y 13, correspondiente a la dislocación de 473 a un precursor bicíclico 474. La elegancia de la estrategia radica en el plan para lograr la ciclación del intermedio bicíclico 474 al esqueleto tetracíclico de la diana 457 en un solo paso. El equivalente sintético 475 de 474 tiene grupos carbonilo adicionales en las posiciones 7 y 9. El primero proporciona activación electrófila adicional en C-7, mientras que el segundo desactiva la nucleofilia del grupo amino. El intermedio bicíclico 475 podría estar disponible a partir de un precursor monocíclico simétrico 476. El grupo amino incipiente de 457, el nitrógeno nitrilo en 476, incluso proporciona activación polar para la construcción de 476 a partir de acrilonitrilo y dihidroresocinal.
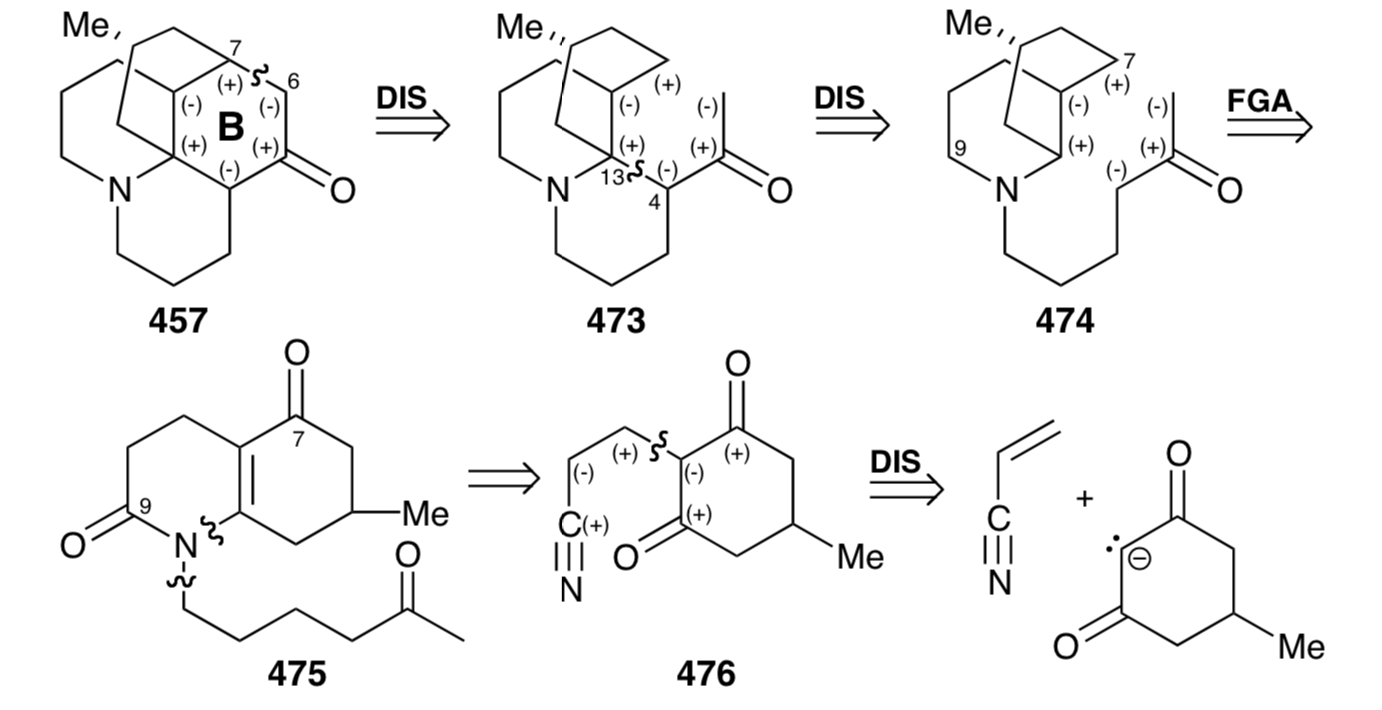
De hecho, la hidrólisis catalizada por ácido de 476 conduce directamente a la enamida 477 que proporcionó 475 por N-alquilación con el bromuro de alquilo 467 y posterior eliminación hidrolítica del grupo cetal enmascarante. La reacción de Michael intramolecular catalizada por bases de 475 podría generar dos estereoisómeros en la posición 13 que resultan de la adición del carbanión a cualquier cara del anillo D en 479. Sin embargo, como se esperaba, el control de enfoque estérico fomenta la adición estereoselectiva en el lado del anillo D opuesto al sustituyente metilo para proporcionar un intermedio 481 en lugar del estereoisómero 480 indeseado. Sin embargo, la síntesis es fatalmente defectuosa debido a que la posterior reacción aldólica dio exclusivamente 484, cuyo esqueleto es epimérico con licopodina en un C12. Así, C-12 en el intermedio 481 es epimerizable, y el epímero 482 aparentemente cicliza en completa preferencia a 481. Esto produce 484 en lugar de 483, que se requiere para la síntesis de licopodina (457). Eliminación reductora de los grupos amida carbonilo e hidroxilo terciario de 484 suministraron 12-epi-licopodina (485).

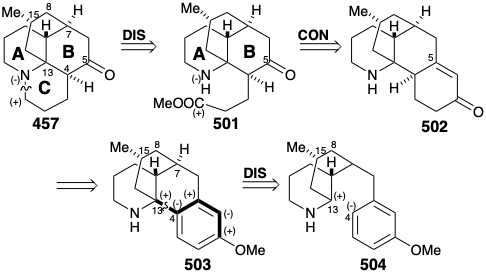
Una estrategia de relevo y un precursor simétrico para licopodina
Una segunda estrategia para la síntesis de licopodina genera el anillo D mediante la ciclación de un sintón tricíclico 486 con anillos AB y C preformados. 20 Esta estrategia fue canalizada por la perspectiva de explotar como material de partida una cetona tricíclica fusionada simétrica 487. Así, el análisis topológico de 457 recomienda la desconexión de dos enlaces entre un átomo común (circular) y uno no común, los enlaces 7-8 y 13-14, para eliminar por completo el anillo D. Se requiere una transposición concomitante del grupo carbonilo de C-5 en 457 a C-6 para generar un precursor simétrico 487. Además, esta transposición en 486 permite la formación del enlace 7-8 C-C del esqueleto de licopodina mediante una alquilación intramolecular que explota la activación polar proporcionada por un carbonilo en la posición 6. Mientras que, el nitrógeno en 486 puede proporcionar activación electrófila para la formación de enlaces C-C en la posición 13 en 487. Se incluye una amida carbonilo en C-9 en 486 como grupo desactivante para disminuir la nucleofilia del grupo amino desfavoreciendo una cuaternización no deseada que podría competir con la alquilación de un nucleófilo carbanión en C7.
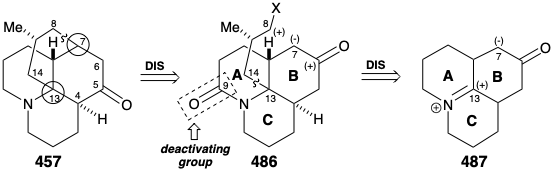
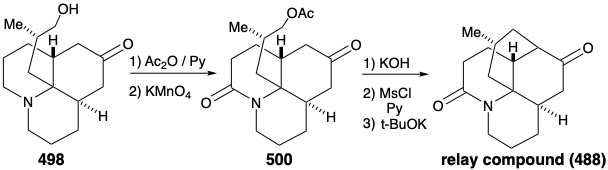
La alquilación intramolecular de la cetona 4 86 produciría 488. Sería tranquilizador que los pasos finales de la síntesis se pudieran elaborar con una muestra de 488 que pudiera prepararse fácilmente a partir del producto natural 457, quizás a través del diol 489, que ya se había preparado a partir de 457 durante estudios estructurales sobre los alcaloides de lycopodium. Esta muestra del compuesto 488 podría entonces ser utilizada, en lugar del material sintético, para elaborar los detalles de la conversión de 488 a 457. Este es otro ejemplo de la estrategia conocida como el enfoque de relevo, que vimos empleado en síntesis de eritronólido B (ver sección 5.4) y quinina (ver sección 6.5). La ventaja de este enfoque es que un valioso intermedio clave se puede obtener fácilmente en cantidad. El compuesto relevador (por ejemplo 488) se convierte en el objetivo de la síntesis.
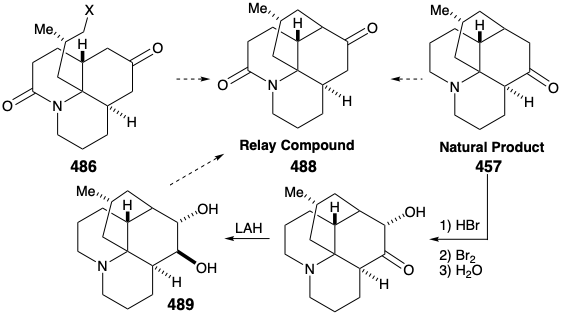
Consideremos primero la interconversión del compuesto relevador 488 y 457 antes de examinar la síntesis total del compuesto relé. Para diferenciar los hidroxilos en las posiciones 5 y 6, el diol 489 de la licopodina natural (457) fue monoacetilado en el hidroxilo C-6 estéricamente más accesible. La deshidratación seguida de hidrólisis proporcionó 490. La oxidación del hidroxilo alílico seguida de la reducción de la cetona α, β-insaturada resultante y la oxidación de permanganato α a la amina terciaria dio el compuesto relevador propuesto, amida 488, en 13% de rendimiento global a partir de licopodina natural (457).
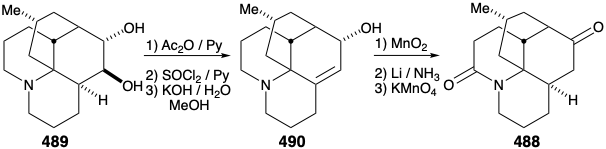
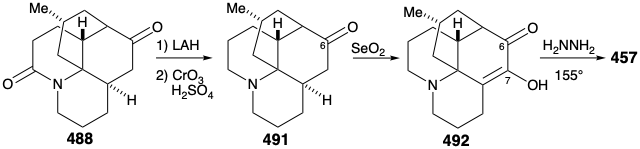
La reconversión del compuesto relevador 488 en licopodina (457) se logró luego mediante la eliminación de la amida carbonilo, mediante la reducción con LAH y la oxidación de los alcoholes epiméricos C-6 resultantes. La aminocetona 491 se oxidó luego al diosfenol 492, que se redujo selectivamente mediante una reacción de Wolff-Kishner con hidrato de hidrazina para proporcionar licopodina (457).
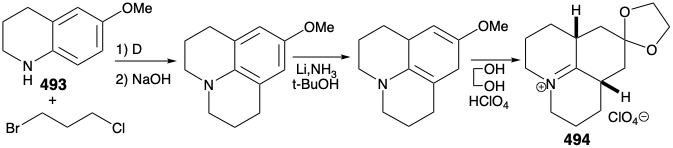
Como se señaló anteriormente, se previó una síntesis de 486, y de ahí el compuesto relevador 488, a partir de un material de partida simétrico (ver arriba). En particular, 486 podría prepararse a partir de 487 por reacción con un sintón nucleofílico de cadena lateral. Con una disposición para enmascarar la electrofilicidad del grupo carbonilo, esta estrategia resultó viable. Así, 494 se preparó a partir de talina (493) por alquilación con 1-bromo-3-cloropropano seguido de disolución de reducción de metales y cetalización. La reacción de 494 con el fragmento nucleofílico 495 seguida de hidrólisis da la amina tricíclica cis, condensada en cis 496. El cierre de anillo de este epíimer es imposible. La epimerización al isómero trans, cis 485 debe preceder al cierre del anillo. Por lo tanto, la epimerización se logró explotando la funcionalidad carbonilo en 496 mediante bromación seguida de deshidrobromación Mattox-Kendall y reducción de metales disueltos. La desmetilación del producto 497 proporcionó entonces una mezcla de cetonas diastereoméricas racémicas 498 y 499. Estos se separaron por cromatografía sobre alúmina. El isómero menor 498 poseía la configuración relativa natural del sustituyente metilo. Así, la presente síntesis no es estereoespecífica, y se produce una pérdida importante casi fatal de material valioso debido a la formación de un estereoisómero 499 no deseado.
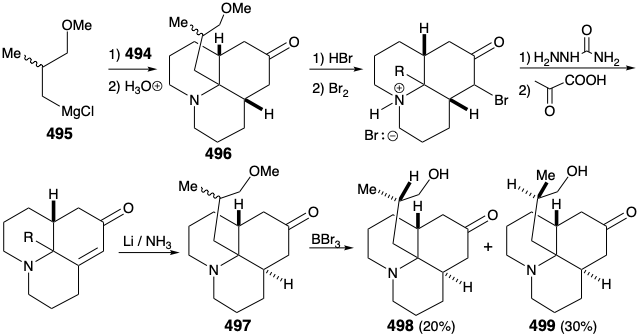
Antes de que se pudiera lograr la alquilación intramolecular de la cetona tricíclica, la nucleofilia del grupo amino tuvo que atenuarse mediante la conversión del grupo amino en 498 en una amida 500 para evitar la N-alquilación. La saponificación, mesilación y alquilación intramolecular proporcionó entonces el compuesto relevador 488 en forma racémica. Dado que 476, aunque homoquirales, derivados de la licopodina natural (457) ya se habían convertido a 457 como se discutió anteriormente, la síntesis total fue completa.
Una estrategia biomimética involuntariamente
En las dos estrategias para licopodina (457) discutidas anteriormente, una generaba el anillo B último y el anillo D el último generado. Ahora consideraremos una estrategia que genere el anillo C último como se encuentra en la biosíntesis de 457. Además, en analogía adicional con la estrategia biosintética, el sustituyente de cadena de tres carbonos en el anillo B de 501, que es el usado para completar el anillo C, se incorpora a un anillo temporal en un precursor 502 por unión al carbono carbonilo (latente) en C-5. Finalmente, el circuito consonante entre los grupos amino y metoxi en el precursor aromático 503 sugiere una construcción polar a partir de 504 del enlace 4-13 C-C, nuevamente en analogía con la estrategia biosintética, por ataque de un electrófilo C-13 sobre un centro nucleófilo en el incipiente C-4 . Sin embargo, esta estrategia se concibió antes de dilucidarse la biosíntesis de licopodina. En palabras del autor de la estrategia, “aunque el plan sintético particular seguido para la construcción del sistema tetracíclico no tenía ninguna base particular en consideraciones biogenéticas, trabajos muy recientes han sugerido una vía biogenética en la que el paso crucial de ciclación es sorprendentemente similar al uno que ideamos”. 21
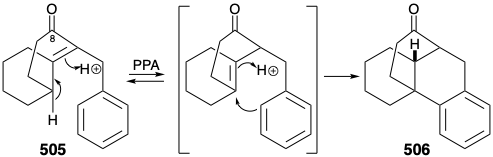
Antes de considerar la implementación exitosa de este plan, es instructivo señalar que se desarrolló con la ayuda de las lecciones aprendidas durante los intentos de lograr una síntesis utilizando otras estrategias fatalmente defectuosas. Un plan temprano infructuoso para una transformación análoga a la generación de 503 a partir de 504 fue apoyado por estudios de modelos exitosos. Así, un modelo carbocíclico 505 se sometió fácilmente a una ciclación análoga a 506 al calentarse con ácido polifosfórico. Se anticipó que el sustituyente metilo en la posición 15 en el anillo D de 504 podría introducirse posteriormente explotando la activación nucleofílica proporcionada por un grupo carbonilo en la posición 8. Además, la inclusión de este grupo carbonilo podría mejorar la utilidad general de la síntesis porque la sustitución de oxígeno se encuentra en la posición 8 en muchos alcaloides de licopodio. Sin embargo, en contraste con el modelo carbocíclico 505, no se pudo lograr la ciclación del análogo heterocíclico 507 para dar 508. Más bien 507 fue aparentemente propenso a la eliminación que conduce a la aromatización.
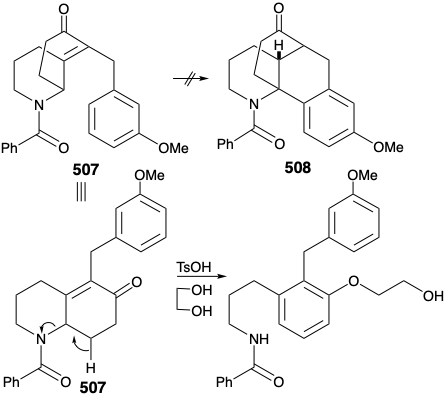
Sin embargo, de este estudio modelo surgió una observación alentadora. Así, el derivado dialquilado 509, un subproducto en la síntesis de 507, sufrió el tipo deseado de ciclación. El éxito de esta ciclación parecía atribuible a dos factores, una orientación axial de un sustituyente bencilo y el bloqueo de la vía de eliminación.
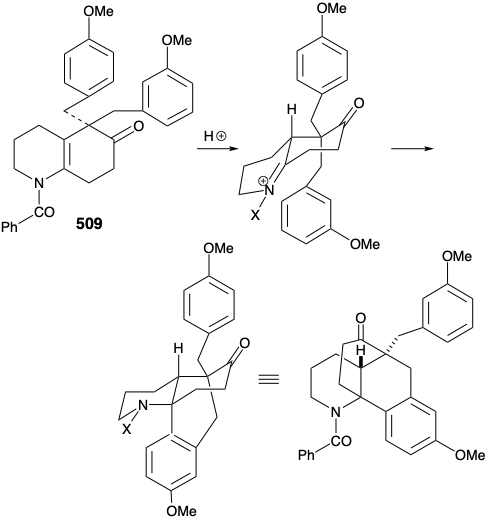
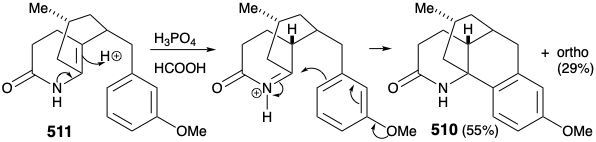
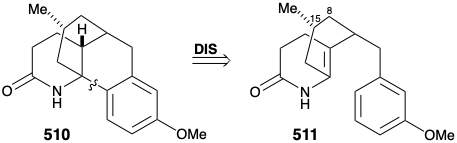
Luego se ideó una estrategia modificada en la que se introdujo el sustituyente metilo requerido en la posición 15 antes de la anelación del anillo B y se eliminó el grupo carbonilo en la posición 8. Además, la estereoquímica de este sustituyente metilo en licopodina dictó una relación trans de los grupos metilo y m-metoxibencilo en la nueva subdiana. El derivado funcionalizado elegido para encarnar estos requisitos fue 511. Se reconoció además que el subtituente de metilo trans en 511 debería eliminar virtualmente la barrera energética para lograr la orientación axial del sustituyente bencilo requerida para la ciclación a 510.
Se exploraron varias estrategias para la síntesis de la subdiana 511, que obviamente se puede derivar de 512. Un acercamiento a 512 mediante la adición conjugada de un nucleófilo de metilo a 513 seguido de la escisión oxidativa de la ciclohexenona 514 fue impedido por la proclividad de 514 a isomerizar en el isómero β, γ-insaturado 515.

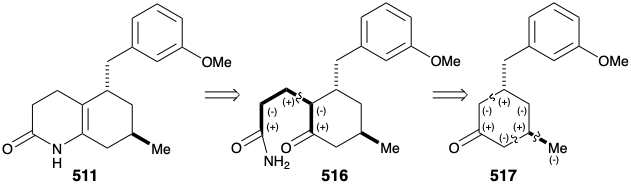
Un segundo acercamiento al 511 explota el circuito consonante entre el carbono carbonilo y el nitrógeno de la enamida en 511 o el circuito consonante relacionado entre los dos grupos carbonilo en la cetoamida 516. La desconexión de la cadena lateral de tres carbonos sugiere un precursor de ciclohexanona 517 que puede ser generado por conexiones polares activadas por el grupo carbonilo.
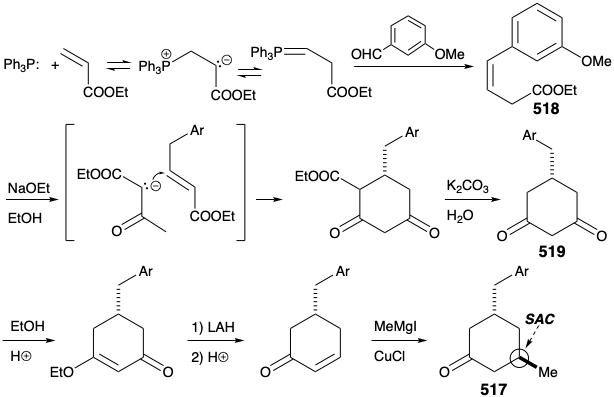
La ciclohexanona 517 se preparó mediante reacciones polares entre un éster acrílico y un éster acetoacético. Así, se generó una diona simétrica 519 a partir del éster β, γ-insaturado 518 por reordenamiento prototrópico alílico, seguido de la adición de Michael de acetoacetato de etilo, ciclación de Dieckmann, hidrólisis y descarboxilación. La reducción selectiva de un solo grupo carbonilo en 519 se facilitó enmascarando el segundo carbonilo de esta diona como un éster vinilógico. La deshidratación catalizada por ácido proporcionó entonces una ciclohexenona que suministró 517 tras la adición estereoselectiva de 1,4-adición de un nucleófilo de metilo.
La estrategia original para la construcción del intermedio clave 511 a partir de ciclohexanona 517 tuvo una deficiencia importante. Así, la conversión de 517 en 511 requiere alquilación regioselectiva de Michael. Sin embargo, la alquilación de 517 ocurrió de manera no regioselectiva en ambos carbonos α al carbonilo produciendo una mezcla de 511 y 520.
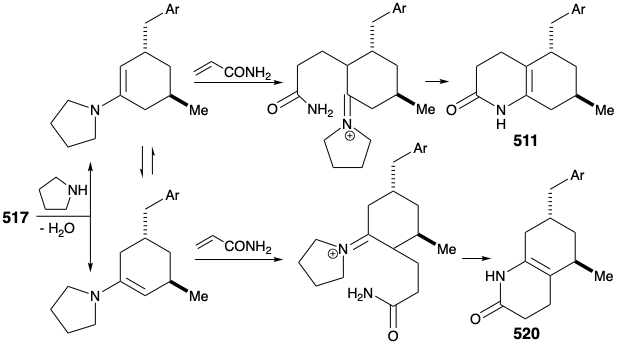
Finalmente se ideó una síntesis mejorada, completamente estructuralmente específica de la subdiana 511 que aprovechó la generación regioespecífica y el atrapamiento electrófilo del enolado 522. Esto se logró mediante la adición de Michael de un nucleófilo bencílico a la ciclohexenona 521. El enolato regioespecífico resultante se alquiló luego con bromuro de alilo. El proceso también es altamente estereoselectivo debido a una preferencia por el ataque axial en las adiciones de Michael de nucleófilos organocobricos y una preferencia por una disposición ecuatorial del sustituyente metilo en 521. Además, la relación trans requerida entre los sustituyentes alilo y bencilo se asegura mediante el control termodinámico debido a la epimerizabilidad α a la cetona carbonilo. La hidroboración y oxidación de la cadena lateral alílica, esterificación y reacción del cetoéster resultante con amoníaco proporcionaron el intermedio clave 511.
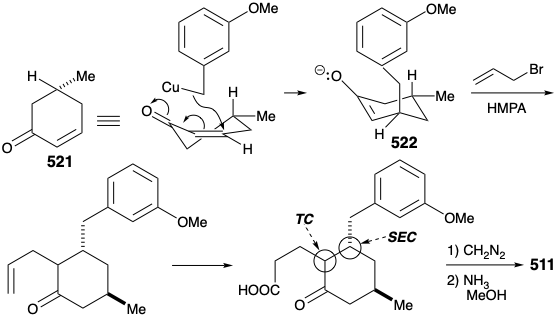
La sustitución aromática electrofílica intramolecular dio principalmente el producto de ciclación para-sustituido deseado 510 (55%) junto con algún producto de sustitución orto (29%). Luego se eliminó la amida carbonilo por reducción con LHA, y la aromaticidad protectora del anillo arilo se eliminó por reducción de Birch. Un plan para efectuar la escisión del anillo por oxidación de una ciclohexenona α, β-insaturada falló debido a una preferencia termodinámica para que la enona 502 requerida existiera como el tautomero 524 β, γ- insaturado correspondiente.
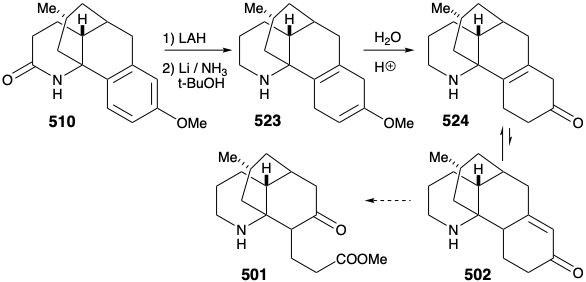
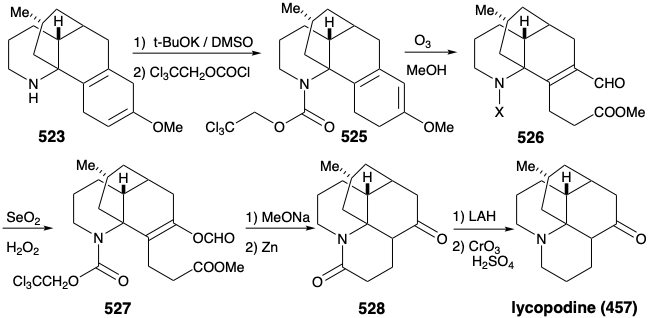
Una vez más, por lo tanto, se tuvo que formular una estrategia alternativa. El plan original para generar 489 a partir de 497 fue fatalmente defectuoso. En efecto, si bien el cada paso en el plan original estaba bien precedido, también lo fue la probabilidad de que 502 estuviera en equilibrio con una cantidad sustancial de 524. En efecto, este mismo problema descarriló un intento de síntesis de 512 de 514 (ver arriba). Como suele ocurrir, una deficiencia de metodología bien conocida para lograr un importante objetivo sintético, sobre todo si impide la conclusión de una síntesis total ambiciosa, inspira la aplicación de la química novedosa para dar solución al dillema. ¡La necesidad es la madre de la invención! Así, la generación de 501 a partir de 523 requirió la escisión oxidativa de dos enlaces, “a” y “b”, en el anillo ciclohexadieno. El plan original requería la escisión del enlace “a” primero después de una isomerización que colocó un enlace C=C fácilmente escindible en esta posición. En la estrategia alternativa, el enlace “b” se escinde primero después de una isomerización que colocó un enlace C=C fácilmente escindible en esta posición.
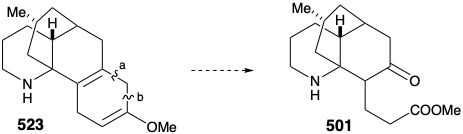
Así, el 1,4-ciclohexadieno 523 se isomerizó a un 1,3-dieno conjugado, y el grupo amino se enmascaró para protegerlo de la oxidación. La ozonólisis selectiva del enlace C=C más rico en electrones en 525 luego proporcionó el éster metílico de aldehído 526. Una inusual oxidación Baeyer-Villager de 526 dio el formiato de enol 527 que proporcionó cetoamida 528 después de metanolisis del éter enólico, eliminación del grupo protector carbamato del nitrógeno amino y lactamización. Luego se eliminó la amida carbonilo para proporcionar licopodina (457) mediante reducción con LAH seguida de reoxidación del hidroxilo C-5 al grupo carbonilo C-5 requerido.