2.4:2.4 Sistemas de enlace Pi conjugado
- Page ID
- 76656
Objetivo de aprendizaje
- reconocer sistemas de enlace pi conjugados
- reconocer que el benceno es aromático
Introducción
Es importante entrenar nuestro ojo para reconocer las características estructurales que tienen efectos estabilizadores. Los enlaces simples y dobles alternantes crean un sistema de enlaces pi conjugados a través de múltiples átomos que reduce la energía y estabiliza la molécula o el ion. Cuando observamos los dobles enlaces carbono-carbono (C=C), necesitamos mirar y ver si están aislados o conjugados.
Para entender la fuente de esta estabilización utilizaremos la teoría de orbitales moleculares (MO). La teoría de los enlaces de valencia hace un trabajo notablemente bueno al explicar la geometría de unión de muchos de los grupos funcionales en compuestos orgánicos, sin embargo, no logra explicar adecuadamente la estabilidad contenida en los enlaces dobles y simples alternantes. Para entender estas propiedades, utilizaremos las ideas de la teoría MO.
Volvamos y consideremos de nuevo el enlace covalente más simple posible: el del hidrógeno molecular (H 2). Cuando describimos la molécula de hidrógeno usando la teoría de enlaces de valencia, dijimos que los dos orbitales de 1 s de cada átomo se superponen, permitiendo que los dos electrones sean compartidos y formando así un enlace covalente. En la teoría orbital molecular, hacemos una declaración adicional: decimos que los dos orbitales atómicos de 1 s se combinan matemáticamente para formar dos nuevos orbitales. Recordemos que un orbital atómico (como el orbital 1s de un átomo de hidrógeno) describe una región del espacio alrededor de un solo átomo dentro del cual es probable que se encuentren electrones. Un orbital molecular describe una región del espacio alrededor de dos o más átomos dentro de los cuales es probable que se encuentren electrones.
Los principios matemáticos nos dicen que cuando los orbitales se combinan, el número de orbitales antes de que se lleve a cabo la combinación debe ser igual al número de nuevos orbitales que resultan de la combinación — ¡los orbitales no desaparecen simplemente! Esto lo vimos anteriormente cuando discutimos orbitales híbridos: uno s y tres p orbitales forman cuatro híbridos sp 3. Cuando dos orbitales atómicos de 1 s se combinan en la formación de H 2, el resultado son dos orbitales sigma (σ).

Orbitales moleculares para H 2
Según la teoría MO, un orbital sigma es menor en energía que cualquiera de los dos orbitales atómicos aislados de 1 s, este orbital sigma inferior se conoce como orbital molecular de enlace. El segundo orbital de la estrella igma es mayor en energía que los dos orbitales atómicos de 1 s, y se conoce como orbital molecular antienlace.
El orbital sigma de unión, que mantiene ambos electrones en el estado fundamental de la molécula, tiene forma de huevo, abarcando los dos núcleos, y con la mayor probabilidad de que los electrones estén en el área entre los dos núcleos. El orbital sigma* antienlace de alta energía se puede visualizar como un par de gotitas, con áreas de mayor densidad de electrones cerca de cada núcleo y un 'nodo', (área de densidad electrónica cero) a medio camino entre los dos núcleos.
Recuerda que aquí estamos pensando en el comportamiento de los electrones como comportamiento de onda. Cuando dos ondas separadas se combinan, pueden hacerlo con interferencia constructiva, donde las dos amplitudes se acumulan y refuerzan entre sí, o interferencia destructiva, donde las dos amplitudes se cancelan entre sí. Los MO de unión son consecuencia de la interferencia constructiva entre dos orbitales atómicos, lo que resulta en una interacción atractiva y un aumento de la densidad de electrones entre los núcleos. Los MO antiligantes son consecuencia de la interferencia destructiva que resulta en una interacción repulsiva y una región de densidad electrónica cero entre los núcleos (es decir, un nodo).
Siguiendo el mismo principio aufbau ('building up') que aprendiste en Química General para escribir configuraciones de electrones, colocamos los dos electrones en la molécula H 2 en el orbital molecular de menor energía, que es el orbital sigma (de unión). El MO de unión (atrayente) está lleno, y el MO antiadhesión (repulsado) está vacío.
Teoría MO y enlaces pi conjugados
La ventaja de usar la teoría MO para entender la unión en moléculas orgánicas se hace más evidente cuando pensamos en enlaces pi. Primero consideremos el enlace pi en eteno desde el punto de vista de la teoría MO (en este ejemplo estaremos desatendiendo los enlaces s en la molécula, y pensando solo en el enlace π). Comenzamos con dos orbitales atómicos: uno orbital 2p no hibridado de cada carbono. Cada uno contiene un solo electrón. En la teoría MO, los dos atómicos se combinan matemáticamente para formar dos orbitales moleculares p i, uno orbital de enlace pi de baja energía y otro orbital antiunión pi* de alta energía.
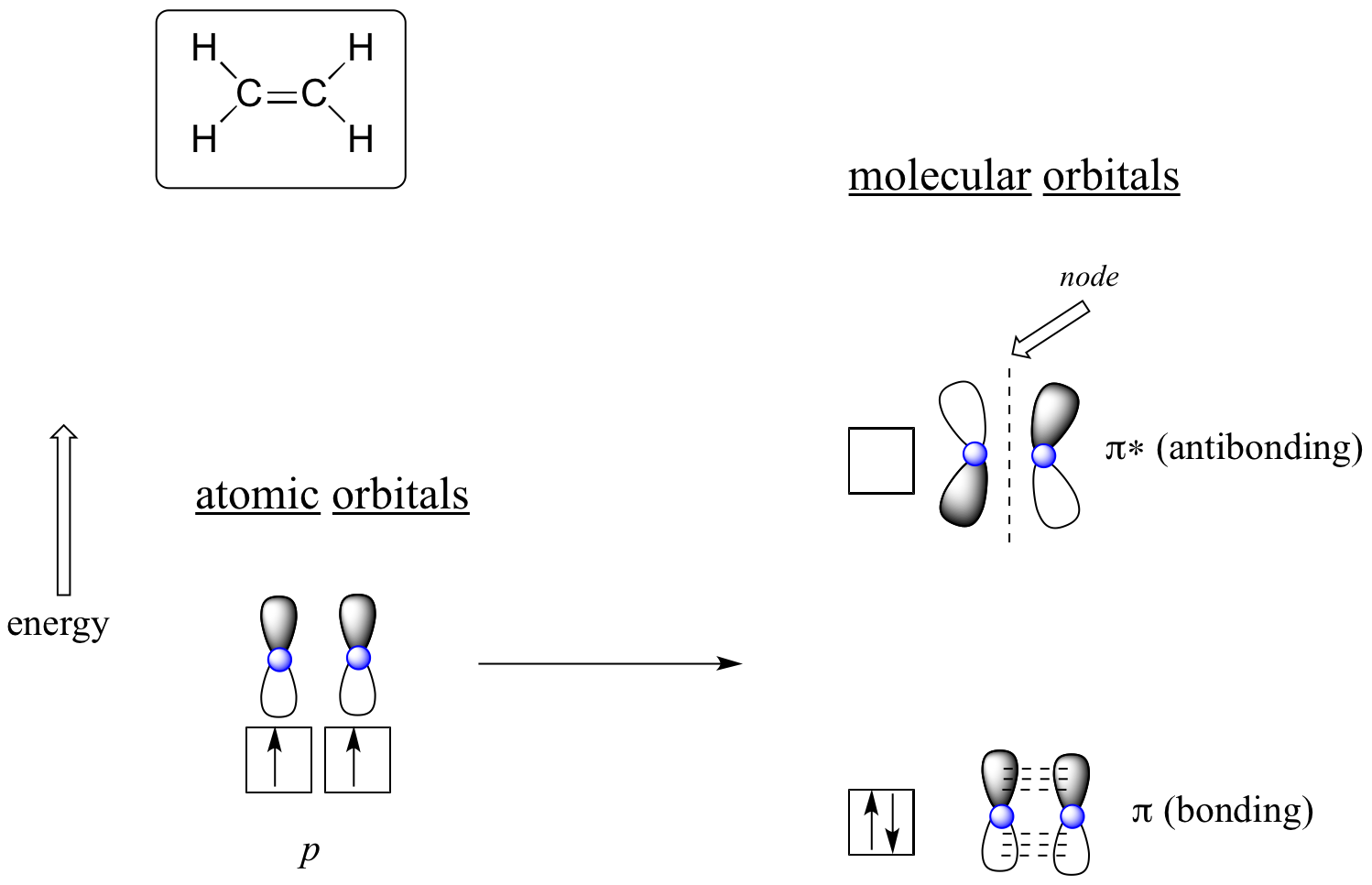
Orbitales moleculares para eteno (etileno)
En el orbital pi de unión, los dos lóbulos sombreados de los orbitales p interactúan constructivamente entre sí, al igual que los dos lóbulos no sombreados (recuerde, la elección de sombreado arbitraria representa signos matemáticos (+) y (-) para la función de onda matemática que describe la órbita). Hay una mayor densidad de electrones entre los dos núcleos de carbono en el orbital molecular, es una interacción de unión.
En el orbital pi* antiadhesión de mayor energía, el lóbulo sombreado de un orbital p interactúa destructivamente con el lóbulo no sombreado del segundo orbital p, conduciendo a un nodo entre los dos núcleos y repulsión general entre los núcleos de carbono.
Nuevamente usando el principio de 'construcción', colocamos los dos electrones en el orbital molecular pi de unión de menor energía. El orbital pi* antiadherentes permanece vacío.
A continuación, consideraremos la molécula de 1,3-butadieno. Solo de la teoría orbital de valencia podríamos esperar que el enlace C 2-C3 en esta molécula, por tratarse de un enlace sigma, pudiera rotar libremente.

Experimentalmente, sin embargo, se observa que existe una barrera significativa a la rotación alrededor del enlace C2-C3, y que toda la molécula es plana. Además, el enlace C2-C3 tiene 148pm de largo, más corto que un enlace sencillo carbono-carbono típico (aproximadamente 154pm), aunque más largo que un doble enlace típico (aproximadamente 134pm).
La teoría orbital molecular da cuenta de estas observaciones con el concepto de enlaces pi deslocalizados. En esta imagen, los cuatro orbitales atómicos 2p se combinan matemáticamente para formar cuatro orbitales moleculares pi de energía creciente. Dos de estos -los orbitales pi de unión- son de menor energía que los orbitales p atómicos a partir de los cuales se forman, mientras que dos -los orbitales pi* antiligantes- son más altos en energía.

El orbital molecular de menor energía, pi 1, solo tiene interacción constructiva y nodos cero. Mayor en energía, pero aún más baja que los orbitales p aislados, el orbital pi 2 tiene un nodo pero dos interacciones constructivas, por lo que sigue siendo un orbital de unión en general. Al observar los dos orbitales antiadhesión, pi 3 * tiene dos nodos y una interacción constructiva, mientras que pi 4 * tiene tres nodos y cero interacciones constructivas.
Por el principio aufbau, los cuatro electrones de los orbitales atómicos aislados de 2 p z se colocan en los MO de unión pi 1 y pi 2. Debido a que pi 1 incluye interacción constructiva entre C 2 y C 3, hay un grado, en la molécula de 1,3-butadieno, de interacción de unión pi entre estos dos carbonos, lo que explica su longitud más corta y la barrera a la rotación. La imagen de enlace de valencia de 1,3-butadieno muestra que los dos enlaces pi están aislados entre sí, con cada par de electrones pi 'pegados' en su propio enlace pi. Sin embargo, la teoría orbital molecular predice (con precisión) que los cuatro electrones pi están en cierta medida deslocalizados, o 'dispersados', sobre todo el sistema pi.

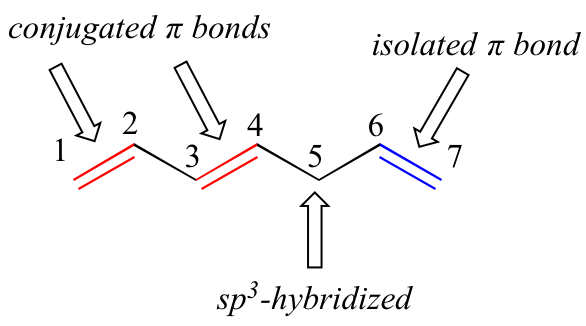
El 1,3-butadieno es el ejemplo más simple de un sistema de enlaces pi conjugados. Para ser considerados conjugados, dos o más enlaces pi deben estar separados por un solo enlace sencillo, es decir, no puede haber un carbono hibridado sp 3 intermedio, porque esto rompería el sistema de superposición de orbitales p paralelos. En el siguiente compuesto, por ejemplo, se conjugan los dobles enlaces C1-C2 y C3-C4, mientras que el doble enlace C 6-C7 se aísla de los otros dos enlaces pi mediante C hibridado sp 3 5.
Un concepto muy importante a tener en cuenta es que existe una estabilidad termodinámica inherente asociada a la conjugación. Esta estabilidad se puede medir experimentalmente comparando el calor de hidrogenación de dos dienos diferentes. (La hidrogenación es un tipo de reacción del que aprenderemos mucho más en el capítulo 15: esencialmente, es el proceso de agregar una molécula de hidrógeno -dos protones y dos electrones- a un enlace p). Cuando los dos dobles enlaces conjugados de 1,3-pentadieno se 'hidrogenan' para producir pentano, se liberan aproximadamente 225 kJ por mol de pentano formado. Compare eso con los aproximadamente 250 kJ/mol liberados cuando se hidrogenan los dos dobles enlaces aislados en 1,4-pentadieno, formando también pentano.
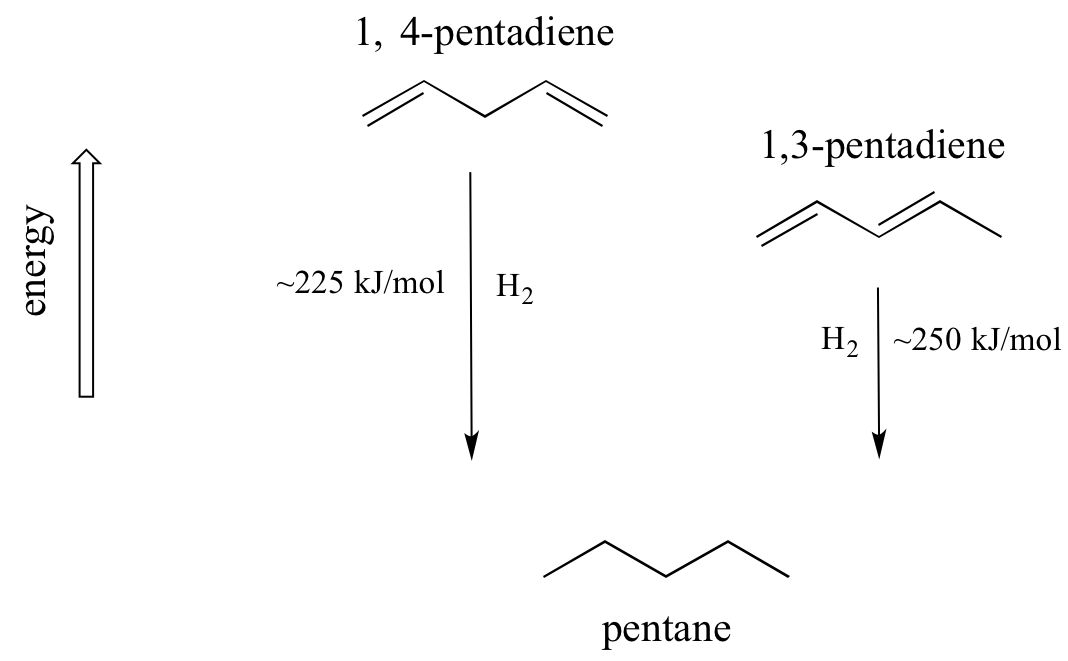
El dieno conjugado es menor en energía: en otras palabras, es más estable. En general, los enlaces pi conjugados son más estables que los enlaces pi aislados.
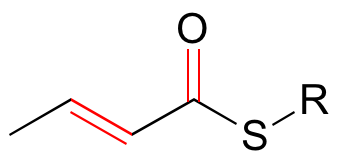
Los sistemas pi conjugados pueden involucrar átomos de oxígeno y nitrógeno, así como carbono. En el metabolismo de las moléculas de grasa, algunas de las reacciones clave involucran alquenos que se conjugan con grupos carbonilo.
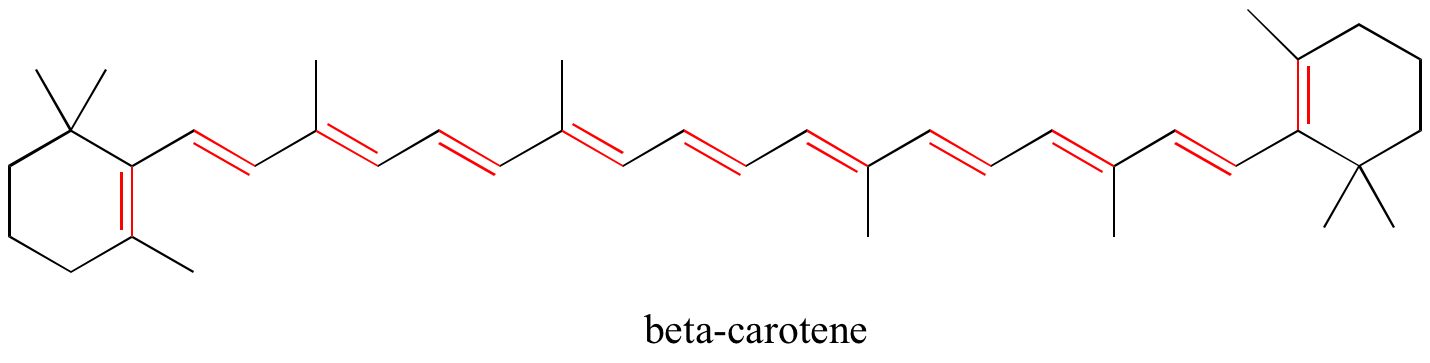
En el capítulo 4, veremos que la teoría MO es muy útil para explicar por qué las moléculas orgánicas que contienen sistemas extendidos de enlaces pi conjugados a menudo tienen colores distintivos. El betacaroteno, el compuesto responsable del color naranja de las zanahorias, tiene un sistema extendido de 11 enlaces pi conjugados.