2.5: Espectroscopia de RMN de Carbono-13




- Última actualización
- 30 oct 2022
- Guardar como PDF

( \newcommand{\kernel}{\mathrm{null}\,}\)
Comenzamos considerando el uso de la espectroscopía deC−13 RMN porque puede proporcionar el tipo de espectro de RMN más simple. C−13es un isótopo menor de carbono, generalmente∼1% de los núcleos de carbono presentes en la muestra sonC−13 (la mayoría sonC−12 y un porcentaje muy pequeño (menos de 1 en un millón sonC−14). En unC−13 espectro, cada átomo de carbono químicamente diferente dará lugar a una señal o pico en el espectro. Por químicamente diferente, queremos decir que los carbonos están en diferentes ambientes; estos ambientes impactan en el campo magnético local que experimentará un núcleo particular. Entonces, por ejemplo, mientras que el etanoCH3CH3 (obviamente) tiene dos carbonos, debido a la simetría de la molécula, ambos núcleos de carbono experimentan el mismo ambiente químico (y magnético local); esperaríamos que el etano muestre un solo pico deC−13 RMN.
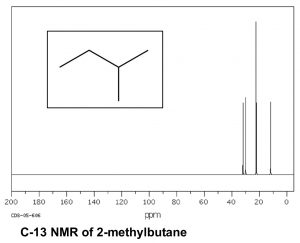
Consideremos ahora un hidrocarburo más complejo como el 2-metilbutano (β), que tiene un total de cinco carbonos; de estos los carbonosC1 yC2 metilo están en ambientes químicos idénticos. La molécula en su conjunto tiene cuatro ambientes distintos y por lo tanto hay 4 picos en su espectro deC−13 RMN.
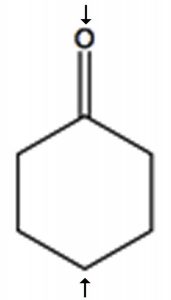
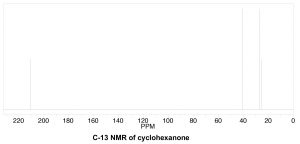
Para determinar el número de carbonos químicamente distintos que están presentes dentro de una molécula, es necesario observar los patrones de enlaces dentro de la molécula, es decir, a qué se une cada carbono. Una forma rápida de verificar entornos químicos idénticos es buscar planos (o ejes) de simetría en la molécula (ignorando las rotaciones alrededor de los enlacesC−C simples). Por ejemplo, la ciclohexanona (\ (\ fila derecha) tiene un solo eje de simetría (marcado por flechas); el resultado es que la molécula tiene solo 4 carbonos químicamente distintos como se muestra en el espectro a continuación. Ahora necesitamos responder a la pregunta de por qué estas señales aparecen en diferentes lugares del espectro
El cambio químico
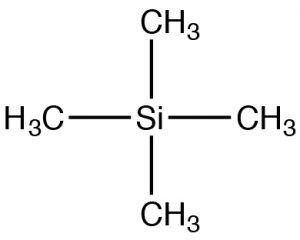
Dado que los instrumentos de RMN pueden tener diferentes potencias de campo, los espectros de RMN a menudo se reportan con referencia a un material estándar: tetrametilsilano (TMS). El TMS tiene simetría tetraédrica, por lo que todos los carbonos de la molécula tienen un ambiente químico idéntico (magnético local). Por lo tanto, podemos predecir con confianza que los espectros deC−13 RMN para TMS tienen un solo pico. El desplazamiento químico (inppm) = desplazamiento de TMS/frecuencia total del espectrómetro. Al utilizar este enfoque, el desplazamiento químico no depende del tipo de instrumento utilizado. ParaC−13 RMN los desplazamientos químicos suelen oscilar entre aproximadamente220 0 y 0. La designación de0 es donde realmente aparecería TMS, aunque esta señal a menudo se elimina del espectro para mayor claridad (como en los dos espectros anteriores). Sin embargo, si ve espectros con un pico a 0 ppm es casi seguro que se deba al estándar TMS y no deben usarse en su determinación de la estructura.
Blindaje y Deshielding
Podemos responder a la pregunta de por qué las señales de RMN aparecen en diferentes lugares recordando primero que no se trata de núcleos aislados, sino de moléculas que consisten en núcleos nucleares rodeados de electrones que pueden describirse como ocupantes de diversos orbitales moleculares. La densidad de electrones alrededor del núcleo tiene un marcado efecto sobre el campo magnético local, es decir, el campo magnético que es “sentido” por cada núcleo. Los electrones también “sienten” el efecto del campo magnético y comienzan a circular alrededor de los núcleos para inducir un nuevo campo magnético que se opone al original. Este impacto global de esto es reducir el campo nuclear efectivo como se muestra en la siguiente figura. Ahora se necesita un campo externo más fuerte para llevar el núcleo a resonancia a la misma frecuencia (el giro del giro nuclear).
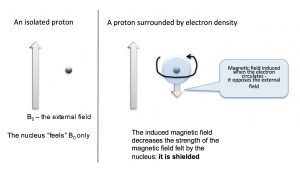
Se dice que los núcleos que están rodeados por una mayor cantidad de densidad de electrones están protegidos; requieren un campo magnético más grande para llevar al giro que los núcleos que están desapantallados (es decir, rodeados de menor densidad de electrones). La absorción por núcleos blindados tiende a ser mayor (“upfield”) en el espectro y los núcleos desblindados aparecen campo abajo. Consideremos por ejemplo, el espectro deC−13 RMN del 1-cloropropano.
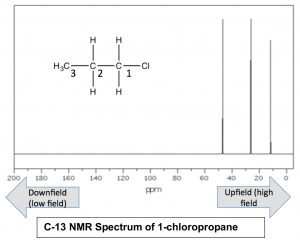
Como cabría esperar, hay 3 picos en el espectro, pero ahora podemos averiguar cuál es cuál, porque podemos predecir las densidades de carga relativas en cada carbono. El carbono unido al cloro (C−1) está más desblindado por el efecto inductivo, y por lo tanto debería aparecer en el campo más bajo. De hecho la señal alrededor47ppm pertenece a laC−1. También podemos ver cómo el efecto inductivo se disipa con la distancia desde el grupo de extracción de electrones hasta laC−3 señal alrededor11ppm. Esto es evidencia directa del efecto inductivo de extracción de electrones.
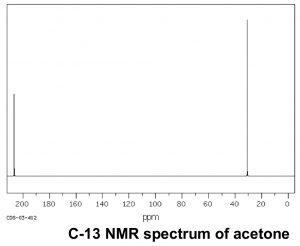
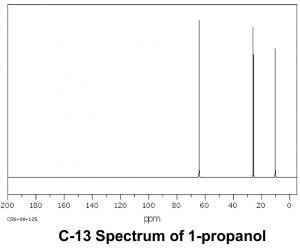
Ahora comparemos este espectro con el de la acetona. Como podríamos predecir, este espectro solo tiene dos picos en él porque la acetona solo tiene dos tipos de carbono, pero lo que es aún más interesante es que elC=O carbono aparece en un campo tan bajo, alrededor205ppm. Esto significa que elC=O carbono debe ser muy deficiente en electrones, mucho más que un carbono unido a un oxígeno (C−O) en un alcohol, que aparece alrededor64ppm (como se muestra en el espectro del 1-propanol). Esto es evidencia directa de un fenómeno que veremos una y otra vez, que es que elC=O carbono es altamente deficiente en electrones y es muy susceptible al ataque nucleofílico. Obsérvese que el espectro de acetona también muestra este pico altamente desplazado hacia abajo correspondiente alC=O carbono.