
II. Reacciones de adición intermolecular
- Page ID
- 79570
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
A. Ecuación general de reacción
Una terminología útil para describir las reacciones de adición de radicales se da en la ecuación 1. Según esta descripción, cuando un radical centrado en carbono reacciona con un doble enlace carbono-carbono, se agrega al átomo de carbono β y crea un nuevo centro radical en el átomo de carbono α. Las letras X, Y y Z en la ecuación 1 representan sustituyentes unidos a los tres átomos de carbono directamente involucrados en la reacción.
.png)
B. Reacción en el átomo de carbono menos sustituido
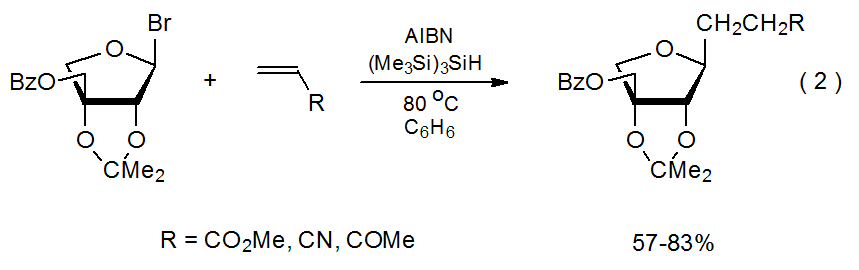
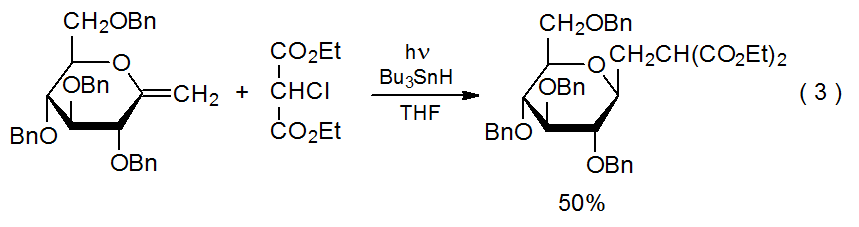
Una característica de las reacciones de adición de radicales es que un radical centrado en carbono se agrega regioselectivamente al átomo menos sustituido en un enlace múltiple C-C. 7—10 La reacción mostrada en el Esquema 1 proporciona un ejemplo típico. Otras reacciones que involucran dobles enlaces con diferentes sustituyentes (eq 2) 11 y dobles enlaces con más de un sustituyente (eq 3) 12.13 exhiben regioselectividad similar. Explicar la regioselectividad en las reacciones de adición comienza señalando que generalmente no son reversibles; 14 por lo tanto, la información sobre las estructuras de estado de transición es crítica para comprender la selectividad en estas reacciones controladas cinéticamente.
.png)
.png)
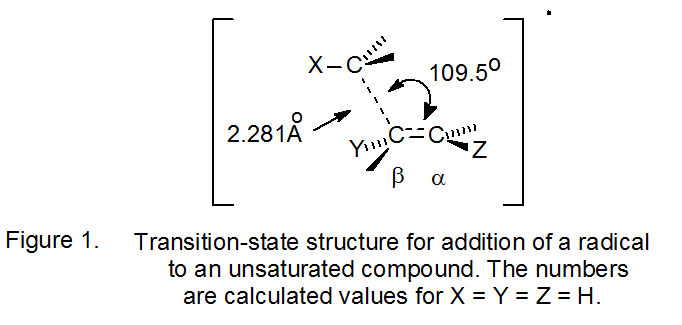
C. Estructura del Estado de Transición
La estructura para el estado de transición en una reacción de adición radical, determinada a partir de cálculos molecular-orbitales, se muestra en la Figura 1. 8 Varios aspectos de esta estructura afectan la regioselectividad de la reacción. La primera es que la estructura es asimétrica. 7,8 Un estado de transición asimétrico requiere que la adición de radicales a cada carbono del enlace múltiple represente una ruta de reacción distinta; no hay un intermedio común. Además, la formación parcial del enlace σ‑entre el átomo de carbono α y el radical entrante, centrado en el carbono, hace que los grupos unidos a cada uno de estos átomos asuman una disposición decididamente piramidal; así, la reacción hace que los grupos unidos a cada centro se acerquen entre sí.
D. Factores que controlan la regioselectividad
La naturaleza asimétrica de la estructura del estado de transición mostrada en la Figura 1 requiere que la adición a cada átomo de carbono de un doble enlace asimétricamente sustituido tenga una constante de velocidad diferente para la reacción. La comprensión de la regioselectividad en las reacciones de adición depende entonces de analizar correctamente los factores que controlan estas dos constantes de velocidad. “La dependencia de temperatura de las constantes de velocidad está bien descrita por la ecuación de Arrhenius k = A exp (- E a/R T). Así, a una temperatura dada, las variaciones de velocidad con sustitución radical y sustrato pueden ser causadas por variaciones en el factor de frecuencia (A) y/o la energía de activación (E a). Para los radicales poliatómicos, los factores de frecuencia abarcan un rango estrecho... De ahí que la gran variación en las constantes de velocidad se deba principalmente a variaciones en la energía de activación”. 8 Los principales factores que determinan la energía de activación [fuerza de unión, efectos estéricos, efectos estereoelectrónicos y efectos polares] son entonces los que deben considerarse para determinar la regioselectividad de reacción. 8
1. Fortalezas de bonos
Una característica de muchas reacciones que son similares en naturaleza es que sus energías de activación (E a) pueden determinarse a partir de la relación Evans—Polanyi (eq 4). 8,10 (La relación Evans—Polanyi se discute en la Sección I.A. del Capítulo 7.) En tales situaciones el cálculo de estas energías depende de determinar las entalpías de reacción (Hr) y establecer valores para las dos constantes en la ecuación 4. Para la adición de radicales centrados en carbono a los dobles enlaces C—C los valores para las constantes determinadas experimentalmente son C=50 kJMol -1 y α=0.25, cuando E a y H r se expresan en kJMol -1. 8,15 El número 0.25 para la constante de proporcionalidad α significa que el cambio de entalpía, que depende de la diferencia en las fortalezas de los enlaces que se rompen y forman, necesita ser grande para que tenga un impacto significativo en la energía de activación para la reacción. El valor de 0.25 para α es razonable para una reacción con un estado de transición temprana.
.png)
2. Efectos Estéricos
La rehibridación del átomo de carbono β de sp 2 a sp 3 se lleva a cabo durante la adición de radicales (eq 1). El necesario reposicionamiento de los grupos que requiere esta rehibridación los fuerza más cercanos (es decir, provoca compresión grupal) a medida que avanza la reacción. Cualquier resistencia a la compresión de grupos causada por impedimentos estéricos eleva la energía requerida para alcanzar el estado de transición para una reacción. 8—10 El estado de transición, por lo tanto, se vuelve energéticamente más difícil de alcanzar a medida que aumenta el tamaño estérico de cualquiera de los grupos unidos al átomo de carbono β. También se produce una compresión estérica similar de los grupos unidos al átomo de carbono que porta el centro radical en el radical aditivo, pero el efecto debe ser menor porque un centro radical típico tiene una estructura que ya se encuentra en una etapa intermedia entre sp 2 y sp 3 hibridación. 8—10
Además de la compresión grupal, las interacciones estéricas en el estado de transición también surgen entre los grupos unidos al átomo de carbono β y los enlazados al radical sumador (Figura 1). El apoyo experimental para la interacción significativa proviene del hallazgo de que las constantes de velocidad para la adición de radicales al átomo de carbono β de un alqueno cambian drásticamente cuando se introducen grupos estéricamente exigentes en este átomo. 7,16 En las reacciones representadas en eq 5 aumentando el tamaño estérico del grupo Y disminuye significativamente la constante de velocidad para la adición β. 7 Si bien puede ser difícil decidir cuánta reducción constante de velocidad es atribuible a la compresión grupal y cuánto a la interacción entre los grupos en el átomo de carbono β y el radical entrante, las constantes de velocidad relativas mostradas en la ecuación 5 dejan pocas dudas de que los efectos estéricos juegan un papel importante en la determinación de las tasas de reacciones de adición de radicales. 7
.png)
Dado que la separación en el estado de transición entre el radical sumador y el átomo de carbono α en una reacción de adición es considerable (Figura 1), es razonable esperar que cualquier obstáculo estérico que involucre sustituyentes α sea pequeño. 7,16 Las constantes de velocidad relativas que se muestran en la ecuación 6 apoyan esta expectativa porque un cambio dramático en el tamaño estérico de los grupos unidos al átomo de carbono α solo tiene un pequeño efecto sobre el valor de estas constantes; la mayor y la más pequeña difieren solo en un factor de 4.2. 7
.png)
Los efectos estéricos tienen un papel más importante en la determinación de la regioselectividad de adición-reacción que las fortalezas de los enlaces que se rompen o forman. La razón de esta situación se remonta a la naturaleza del proceso de adición. En las reacciones competitivas que determinan la regioselectividad [es decir, la adición al átomo de carbono α o β en un enlace múltiple de un compuesto insaturado] se están rompiendo y formando el mismo número y tipos de enlaces; en consecuencia, debería haber poca diferencia en las energías de activación para estos dos reacciones basadas solo en las fuerzas de unión.
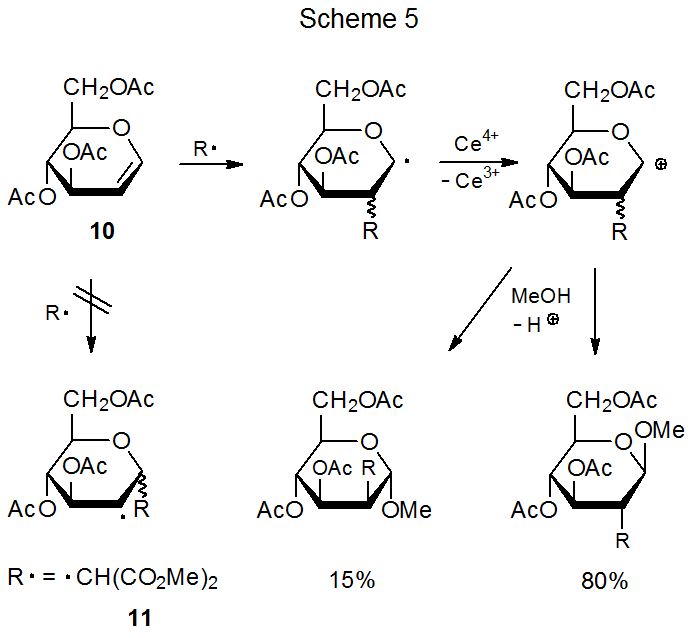
Aunque el papel principal de los efectos estéricos en la determinación de la regioselectividad en las reacciones de adición radical es claro, estos efectos no siempre son el único factor determinante. Sería difícil, por ejemplo, explicar la adición preferencial a C-2 en el glical 10 (Esquema 5) sobre la base solo de los efectos estéricos porque C-2 está, en todo caso, más obstaculizado que C-1. 17—19 Claramente, otro factor también afecta la regioselectividad en reacciones de este tipo.
3. Efectos polares
Los efectos polares son influencias sobre la reactividad causadas por la distribución desigual de electrones dentro de una molécula o intermedio reactivo. En las reacciones de adición radical estos efectos pueden originarse con grupos sustituyentes y pueden transmitirse a los átomos que reaccionan ya sea a través de enlaces o a través del espacio. Los efectos polares también pueden surgir de la deslocalización de electrones que produce una distribución desigual de electrones.
Los datos mostrados en la ecuación 7 ilustran la importancia que tienen los efectos polares en las reacciones de adición de radicales. 7 Estos datos describen las constantes de velocidad relativas para la adición del radical ciclohexilo nucleófilo (C 6 H 11 ·) a ésteres sustituidos α, β-insaturados. La constante de velocidad es grande cuando un sustituyente fuertemente aceptor de electrones (por ejemplo, CN, CO 2 Me) está unido al átomo de carbono α en uno de estos ésteres insaturados. La extracción de electrones del doble enlace por CN o CO 2 Me se debe principalmente a la deslocalización que desplaza la densidad de electrones a uno de estos sustituyentes α. En el caso de que el sustituyente α-sea un grupo metoxicarbonil (Z = CO 2 Me), el desplazamiento de densidad de electrones se puede observar en las estructuras de resonancia contribuyentes que se muestran en la Figura 2. Aunque los efectos polares que se describen son los que existen en los reactivos, las constantes de velocidad en eq 7 apoyan la idea de que estos efectos siguen siendo significativos en el estado de transición.
.png)
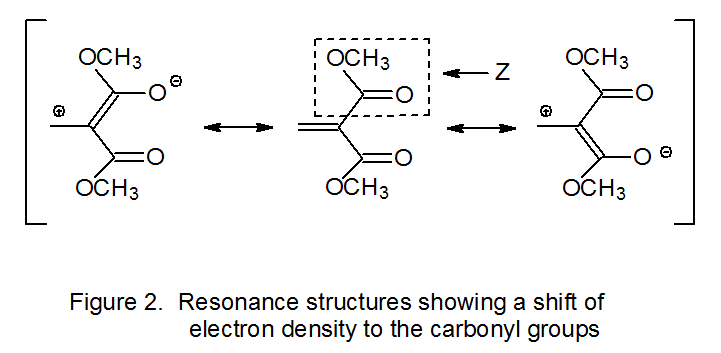
Los efectos polares no solo explican la diferencia en las constantes de velocidad para las reacciones mostradas en la ecuación 7 sino que también racionalizan la regioselectividad de estas reacciones. El híbrido de resonancia representado en la Figura 2 indica una densidad electrónica reducida en el átomo β-carbono en el doble enlace carbono-carbono del éster; en consecuencia, este átomo representa un punto de atracción para un radical nucleófilo. En tal situación regioselectiva, se puede esperar la adición de β-átomos de carbono. Un ejemplo de este tipo de adición se muestra en el Esquema 6 donde el radical carbohidrato nucleofílico 13 se suma regioselectivamente al átomo de carbono β de la cetona α, β-insaturada 12. 20

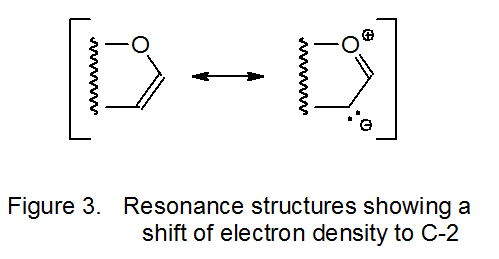
La adición de un radical nucleofílico a un doble enlace rico en electrones es demasiado lenta para competir con otras reacciones radicales, pero si el radical es electrófilo, se produce la adición. El radical dimetilmalonilo 11, por ejemplo, se suma al doble enlace rico en electrones en el D-glucal 10 (Esquema 5). 17—19 Como indica el híbrido de resonancia representado en la Figura 3, C-2 en 10 tiene mayor densidad de electrones que C-1; así, el radical electrófilo 11 no solo se suma al doble enlace en 10 sino que lo hace regioselectividad en C-2 (Esquema 5).
Dado que los efectos estéricos y polares a menudo favorecen la formación del mismo producto en una reacción de adición radical (es decir, que desde la adición al átomo de carbono menos sustituido en el doble enlace del reactivo insaturado), a veces es difícil determinar la contribución relativa de cada efecto a la regioselectividad de una reacción. Una serie de experimentos diseñados para probar estas contribuciones se muestra en la ecuación 8. 7 El primer experimento implica la adición del radical ciclohexilo al acrilato de metilo (eq 8, R = H). En esta reacción, tanto los efectos estéricos como los polares favorecen la adición del radical ciclohexilo nucleofílico al átomo de carbono menos sustituido en el doble enlace carbono-carbono, pero a medida que el grupo R se vuelve estéricamente mayor, la regioselectividad de la reacción disminuye. Para el grupo R estéricamente más grande la dirección de adición favorecida realmente cambia. El mensaje aquí es que los efectos estéricos pueden abrumar los efectos polares al establecer la regioselectividad de reacción, pero un grupo estéricamente bastante exigente (por ejemplo, un grupo t-butilo) es necesario para superar un fuerte efecto polar.
.png)
Otra indicación de la significación de los efectos polares en las reacciones de adición radical se puede observar volviendo a la relación Evans-Polanyi (eq 4). Esta relación se aplica a las reacciones de adición radical en las que los factores polares no son importantes. Para las reacciones donde los factores polares son importantes, las energías de activación son menores que las calculadas a partir de la ecuación 4. En tales situaciones es más precisa una ecuación modificada (eq 9), una que incluye los términos multiplicativos F n y F e, reflejando efectos polares nucleofílicos y electrofílicos, respectivamente. 8,15
.png)
4. Interacciones Fronteria-Orbital
Debido a que las reacciones de adición radical tienen estados de transición temprana, 7 interacciones frontera-orbital son capaces de proporcionar un enfoque alternativo para explicar la regioselectividad de reacción. El primer paso en este enfoque es identificar los orbitales fronterizos en la reacción de interés; por ejemplo, en la adición del radical dimetilmalonilo 11 al doble enlace rico en electrones en el D-glucal 10 (Esquema 5), la interacción primaria es entre el SOMO de 11 y el HOMO de 10 (Figura 10 en el Capítulo 7). Identificar las interacciones frontera-orbital en una reacción no explica, por sí misma, la regioselectividad de reacción, pero la identificación orbital es un primer paso crítico para tal comprensión porque de los orbitales fronterizos provienen los coeficientes atómico-orbitales que forman la base para explicar regio selectividad.
Los coeficientes orbitales atómicos son valiosos para determinar la regioselectividad de una reacción con un estado de transición temprano porque la constante de velocidad para la reacción de formación de enlaces entre dos átomos en tal reacción depende en gran medida de la magnitud de los coeficientes en su frontera de interacción orbitales. 17,18 En la reacción que se muestra en el Esquema 5 el enlace más efectivo es entre el radical 11 y C-2 en el D-glucal 10 porque el coeficiente orbital atómico en C-2 para el HOMO en 10 es mayor que el de C‑1 (Figura 4); 21 en consecuencia, se favorece la adición regioselectiva a C-2. 17,18
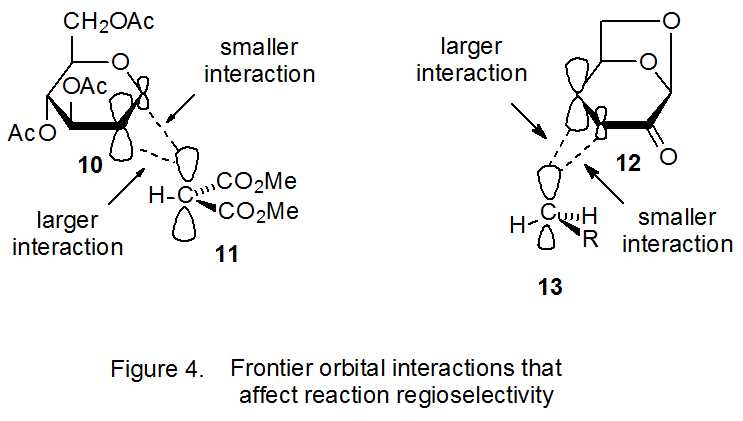
Las interacciones frontera-orbital también explican la regioselectividad en la reacción de adición mostrada en el Esquema 6, donde un radical nucleofílico (13) se está agregando a un doble enlace deficiente en electrones. 20 La adición del radical 13 a C‑4, en lugar de C-3, en la cetona α, β-insaturada 12 no puede explicarse por efectos estéricos, pero las interacciones frontera-orbital sí proporcionan una base para comprender la regioselectividad observada. La interacción más importante en este caso es entre el SOMO de 13 y el LUMO de 12. (Una justificación para ello es la interacción primaria, frontera-orbital se da en la Sección IV.B.1 del Capítulo 7) Para una LUMO como la de 12 el mayor coeficiente orbital atómico se asocia con el orbital p en C-4 (Figura 4); 21 consecuentemente, addi regioselectivo ción a C-4 es favorecida. 22
5. Aducto-Estabilización Radical
La estabilización aducto-radical como una posibilidad para explicar la adición regioselectiva de un radical centrado en carbono a un enlace múltiple no es muy apreciada porque la naturaleza exotérmica y el probable estado de transición temprana para las reacciones de adición radical argumentan en contra del estado de transición significativo estabilización por el radical en desarrollo. La evidencia del estudio de compuestos modelo que se cita en apoyo de este punto de vista es que el radical ciclohexilo se agrega más rápidamente a la acroleína y al acrilonitrilo que al estireno (eq 10), 7,23 a pesar de que un grupo fenilo es más efectivo para estabilizar un centro radical que un carbonilo o grupo ciano. 7,24 Esta información indica que la estabilización aducto-radical es menos importante que los efectos polares en el estado de transición para una reacción de adición; por lo tanto, los efectos polares son los principales responsables de las diferencias en la reactividad de los compuestos insaturados, diferencias como las mostradas en eq 10. 7 (Como se discute en la Sección II.D.2 y se ve en la ecuación 6, el impedimento estérico de los sustituyentes α utilizados en las reacciones mostradas en la ecuación 10 debe ser intrascendente.) Dado que las reacciones entre alquenos deficientes en electrones y radicales nucleofílicos se estabilizan en el estado de transición por efectos polares, estos efectos podrían enmascarar menos importante, la estabilización aducto-radical. Una mejor prueba de la importancia de la estabilización aducto-radical en la adición regioselectiva sería aquella en la que los efectos polares no pudieran ser el factor determinante.
.png)
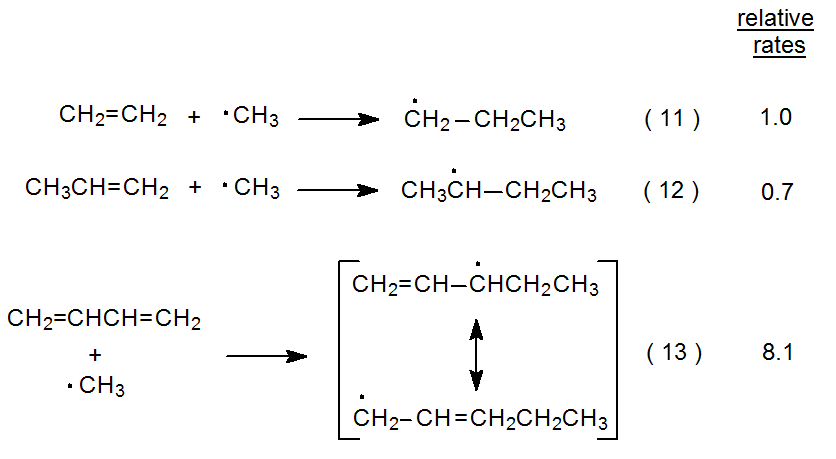
Las reacciones de adición mostradas en las ecuaciones 11 y 12 son aquellas para las que los efectos polares deben ser mínimos. 10 La similitud en las tasas relativas para las dos reacciones indica que la estabilización aducto-radical es intrascendente en el estado de transición. Estas reacciones también subrayan la dificultad de eliminar completamente la influencia de los efectos polares al comparar reacciones radicales. La velocidad ligeramente reducida para la reacción mostrada en la ecuación 12, cuando se compara con la de la ecuación 11, podría deberse al efecto del grupo metilo débilmente donador de electrones en el propeno reduciendo en pequeña medida la velocidad de adición de un radical nucleofílico a un doble enlace ligeramente más rico en electrones. (Como se menciona en el Capítulo 7, Secciones III.C. y III.E., no existe un acuerdo completo sobre la nucleofilia del radical metilo.)
-(13).png)
La reacción mostrada en la ecuación 13 apoya la idea de que la estabilidad del radical en desarrollo puede ser un factor en la reducción de la energía del estado de transición. 10 La mayor velocidad para esta reacción, en comparación con las mostradas en las ecuaciones 11 y 12, puede explicarse por la estabilización de resonancia en el radical en desarrollo contribuyendo significativamente a la estabilización del estado de transición. Los datos limitados en las ecuaciones 11-13 concuerdan con la idea de que la estabilidad aducto-radical es solo un factor en las reacciones de adición de radicales cuando dicha estabilización es considerable. Una vez más, sin embargo, los efectos polares nublan esta interpretación. El 1,3-butadieno puede verse como una molécula en la que cada doble enlace tiene un sustituyente etenilo unido. Dicho sustituyente debería ser aceptor de electrones o, al menos, menos donador de electrones que un grupo metilo; en consecuencia, los dobles enlaces en 1,3-butadieno deberían ser más reactivos hacia el radical metilo que el doble enlace en propeno. Esta diferencia podría explicar, al menos en parte, la diferencia en las velocidades relativas para las reacciones mostradas en la ecuación 12 y la ecuación 13.