
7.7: Extracciones Líquido-Líquido
- Page ID
- 75953

La extracción líquido-líquido es una importante técnica de separación para laboratorios ambientales, clínicos e industriales. Un método estándar de análisis ambiental ilustra la importancia de las extracciones líquido-líquido. Los departamentos municipales de agua monitorean rutinariamente los suministros públicos de agua para detectar trihalometanos (CHCl 3, ChBrCl 2, ChBr 2 Cl y ChBr 3) porque se conocen o sospechan carcinógenos. Antes de su análisis por cromatografía de gases, los trihalometanos se separan de su matriz acuosa mediante una extracción líquido-líquido con pentano [“El análisis de trihalometanos en agua potable por extracción líquida”, ePamethod501.2 (EPA 500-Series, noviembre de 1979)].
La Agencia de Protección Ambiental (EPA) también publica dos métodos adicionales para los trihalometanos. Los métodos 501.1 y 501.3 utilizan una purga y trampa para recolectar los trihalometanos antes de un análisis cromatográfico de gases con un detector específico de haluros (Método 501.1) o un espectrómetro de masas como detector (Método 501.3). Encontrará más detalles sobre la cromatografía de gases, incluyendo detectores, en el Capítulo 12.
En una simple extracción líquido-líquido, el soluto se divide entre dos fases inmiscibles. Una fase suele ser un disolvente acuoso y la otra fase es un disolvente orgánico, tal como el pentano utilizado para extraer trihalometanos del agua. Debido a que las fases son inmiscibles forman dos capas, con la fase más densa en el fondo. El soluto inicialmente está presente en una de las dos fases; después de la extracción está presente en ambas fases. La eficiencia de extracción, es decir, el porcentaje de soluto que se mueve de una fase a otra, está determinada por la constante de equilibrio para la partición del soluto entre las fases y cualquier otra reacción secundaria que involucre al soluto. Ejemplos de otras reacciones que afectan la eficiencia de extracción incluyen reacciones ácido-base y reacciones de complejación.
Coeficientes de partición y relaciones de distribución
Como aprendimos anteriormente en este capítulo, la partición de un soluto entre dos fases se describe mediante un coeficiente de partición, K D. Si extraemos un soluto de una fase acuosa a una fase orgánica
\[S_{a q} \rightleftharpoons S_{o r g} \nonumber\]
entonces el coeficiente de partición es
\[K_{\mathrm{D}}=\frac{\left[S_{org}\right]}{\left[S_{a q}\right]} \nonumber\]
Un gran valor para K D indica que la extracción de soluto a la fase orgánica es favorable.
Para evaluar la eficiencia de una extracción debemos considerar la concentración total del soluto en cada fase, la cual definimos como una relación de distribución, D.
\[D=\frac{\left[S_{o r g}\right]_{\text { total }}}{\left[S_{a q}\right]_{\text { total }}} \nonumber\]
El coeficiente de partición y la relación de distribución son idénticos si el soluto tiene solo una forma química en cada fase; sin embargo, si el soluto existe en más de una forma química en cualquiera de las fases, entonces K D y D suelen tener valores diferentes. Por ejemplo, si el soluto existe en dos formas en la fase acuosa, A y B, solo una de las cuales, A, se divide entre las dos fases, entonces
\[D=\frac{\left[S_{o r g}\right]_{A}}{\left[S_{a q}\right]_{A}+\left[S_{a q}\right]_{B}} \leq K_{\mathrm{D}}=\frac{\left[S_{o r g}\right]_{A}}{\left[S_{a q}\right]_{A}} \nonumber\]
Esta distinción entre K D y D es importante. El coeficiente de partición es una constante de equilibrio termodinámico y tiene un valor fijo para la partición del soluto entre las dos fases. El valor de la relación de distribución, sin embargo, cambia con las condiciones de la solución si cambian las cantidades relativas de A y B. Si conocemos las reacciones de equilibrio del soluto dentro de cada fase y entre las dos fases, podemos derivar una relación algebraica entre K D y D.
Extracción líquido-líquido sin reacciones secundarias
En una simple extracción líquido-líquido, la única reacción que afecta la eficiencia de extracción es la partición del soluto entre las dos fases (Figura 7.7.1 ).
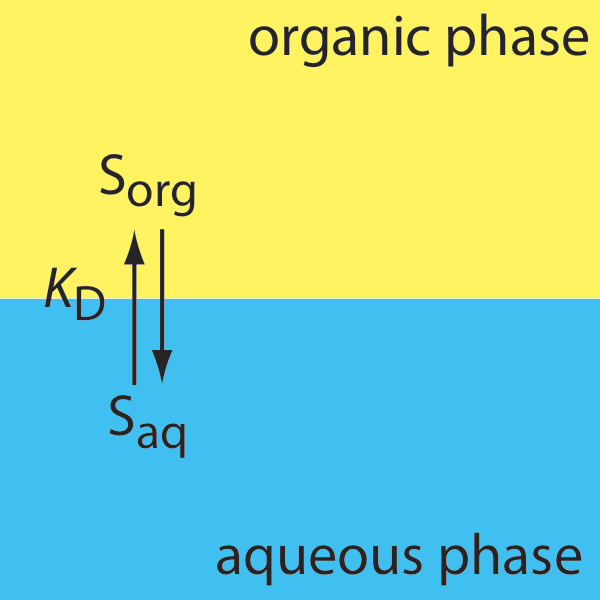
En este caso la relación de distribución y el coeficiente de partición son iguales.
Supongamos que el soluto inicialmente está presente en la fase acuosa y que deseamos extraerlo en la fase orgánica. Una conservación de la masa requiere que los moles de soluto inicialmente presentes en la fase acuosa sean iguales a los moles combinados de soluto en la fase acuosa y la fase orgánica después de la extracción.
donde los subíndices indican el número de extracción con 0 representando el sistema antes de la extracción y 1 el sistema después de la primera extracción. Después de la extracción, la concentración de soluto en la fase acuosa es
y su concentración en la fase orgánica es
donde V ac y V org son los volúmenes de la fase acuosa y la fase orgánica. Resolviendo la Ecuación\ ref {7.2} para (mol S org) 1 y sustituyendo en la Ecuación\ ref {7.4} déjanos con
Sustituyendo la ecuación\ ref {7.3} y la ecuación\ ref {7.5} en la ecuación\ ref {7.1} da
\[D = \frac {\frac {(\text{mol }S_{aq})_0-(\text{mol }S_{aq})_1} {V_{org}}} {\frac {(\text{mol }S_{aq})_1} {V_{aq}}} = \frac{\left(\operatorname{mol} \ S_{a q}\right)_{0} \times V_{a q}-\left(\operatorname{mol} \ S_{a q}\right)_{1} \times V_{a q}}{\left(\operatorname{mol} \ S_{a q}\right)_{1} \times V_{o r g}} \nonumber\]
Reordenando y resolviendo la fracción de soluto que permanece en la fase acuosa después de una extracción, (q ac) 1, da
La fracción presente en la fase orgánica después de una extracción, (q org) 1, es
\[\left(q_{o r g}\right)_{1}=\frac{\left(\operatorname{mol} S_{o r g}\right)_{1}}{\left(\operatorname{mol} S_{a q}\right)_{0}}=1-\left(q_{a q}\right)_{1}=\frac{D V_{o r g}}{D V_{o r g}+V_{a q}} \nonumber\]
El ejemplo 7.7.1 muestra cómo podemos usar la Ecuación\ ref {7.6} para calcular la eficiencia de una simple extracción líquido-líquido.
Un soluto tiene un K D entre agua y cloroformo de 5.00. Supongamos que extraemos una muestra de 50.00-mL de una solución acuosa 0.050 M del soluto usando 15.00 mL de cloroformo. a) ¿Cuál es la eficiencia de extracción de la separación? b) ¿Qué volumen de cloroformo necesitamos si queremos extraer el 99.9% del soluto?
Solución
Para una extracción simple líquido-líquido, la relación de distribución, D, y el coeficiente de partición, K D, son idénticos.
(a) La fracción de soluto que permanece en la fase acuosa después de la extracción viene dada por la Ecuación\ ref {7.6}.
\[\left(q_{aq}\right)_{1}=\frac{V_{a q}}{D V_{org}+V_{a q}}=\frac{50.00 \ \mathrm{mL}}{(5.00)(15.00 \ \mathrm{mL})+50.00 \ \mathrm{mL}}=0.400 \nonumber\]
La fracción de soluto en la fase orgánica es 1—0.400, o 0.600. La eficiencia de extracción es el porcentaje de soluto que se mueve a la fase de extracción; por lo tanto, la eficiencia de extracción es de 60.0%.
(b) Para extraer 99.9% del soluto (q aq) 1 debe ser 0.001. Resolviendo la ecuación\ ref {7.6} para V org, y haciendo sustituciones apropiadas para (q aq) 1 y V aq da
\[V_{o r g}=\frac{V_{a q}-\left(q_{a q}\right)_{1} V_{a q}}{\left(q_{a q}\right)_{1} D}=\frac{50.00 \ \mathrm{mL}-(0.001)(50.00 \ \mathrm{mL})}{(0.001)(5.00 \ \mathrm{mL})}=999 \ \mathrm{mL} \nonumber\]
Se trata de un gran volumen de cloroformo. Claramente, una sola extracción no es razonable bajo estas condiciones.
En Example 7.7.1 , una sola extracción proporciona una eficiencia de extracción de solo 60%. Si realizamos una segunda extracción, la fracción de soluto que queda en la fase acuosa, (q aq) 2, es
\[\left(q_{a q}\right)_{2}=\frac{\left(\operatorname{mol} \ S_{a q}\right)_{2}}{\left(\operatorname{mol} \ S_{a q}\right)_{1}}=\frac{V_{a q}}{D V_{org}+V_{a q}} \nonumber\]
Si V aq y V org son iguales para ambas extracciones, entonces la fracción acumulada de soluto que permanece en la capa acuosa después de dos extracciones, (Q ac) 2, es el producto de (q aq) 1 y (q aq) 2, o
\[\left(Q_{aq}\right)_{2}=\frac{\left(\operatorname{mol} \ S_{aq}\right)_{2}}{\left(\operatorname{mol} \ S_{aq}\right)_{0}}=\left(q_{a q}\right)_{1} \times\left(q_{a q}\right)_{2}=\left(\frac{V_{a q}}{D V_{o r g}+V_{a q}}\right)^{2} \nonumber\]
En general, para una serie de n extracciones idénticas, la fracción de analito que permanece en la fase acuosa después de la última extracción es
\[\left(Q_{a q}\right)_{n}=\left(\frac{V_{a q}}{D V_{o r g}+V_{a q}}\right)^{n} \label{7.7}\]
Para la extracción descrita en el Ejemplo 7.7.1 , determinar (a) la eficiencia de extracción para dos extracciones idénticas y para tres extracciones idénticas; y (b) el número de extracciones requeridas para asegurar que extraemos 99.9% del soluto.
Solución
(a) La fracción de soluto restante en la fase acuosa después de dos extracciones y tres extracciones es
\[\left(Q_{aq}\right)_{2}=\left(\frac{50.00 \ \mathrm{mL}}{(5.00)(15.00 \ \mathrm{mL})+50.00 \ \mathrm{mL}}\right)^{2}=0.160 \nonumber\]
\[\left(Q_{a q}\right)_{3}=\left(\frac{50.0 \ \mathrm{mL}}{(5.00)(15.00 \ \mathrm{mL})+50.00 \ \mathrm{mL}}\right)^{3}=0.0640 \nonumber\]
Las eficiencias de extracción son 84.0% para dos extracciones y 93.6% para tres extracciones.
(b) Para determinar el número mínimo de extracciones para una eficiencia de 99.9%, establecemos (Q aq) n en 0.001 y resolvemos para n usando la Ecuación\ ref {7.7}.
\[0.001=\left(\frac{50.00 \ \mathrm{mL}}{(5.00)(15.00 \ \mathrm{mL})+50.00 \ \mathrm{mL}}\right)^{n}=(0.400)^{n} \nonumber\]
Tomando el tronco de ambos lados y resolviendo para n
\[\begin{aligned} \log (0.001) &=n \log (0.400) \\ n &=7.54 \end{aligned} \nonumber\]
encontramos que es necesario un mínimo de ocho extracciones.
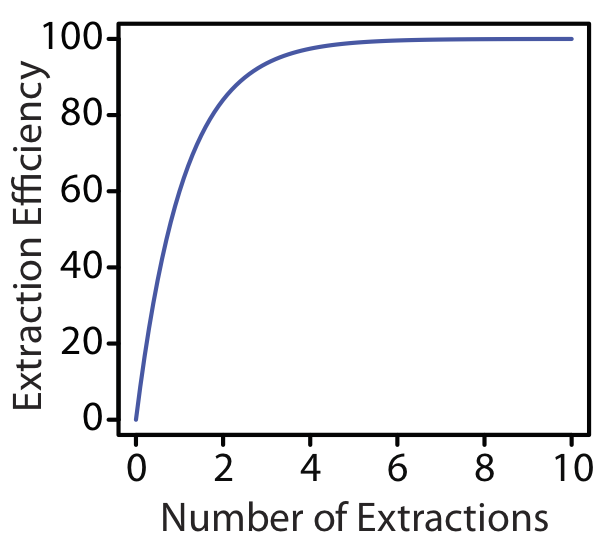
Los dos últimos ejemplos nos proporcionan una observación importante: para cualquier eficiencia de extracción, necesitamos menos solvente si completamos varias extracciones usando porciones más pequeñas de solvente en lugar de una extracción usando un mayor volumen de solvente. Para las condiciones del Ejemplo 7.7.1 y Ejemplo 7.7.2 , una eficiencia de extracción de 99.9% requiere una extracción con 9990 mL de cloroformo, o 120 mL cuando se usan ocho porciones de 15 mL de cloroformo. Aunque la eficiencia de extracción aumenta drásticamente con los primeros múltiples, el efecto disminuye rápidamente a medida que aumentamos el número de extracciones (Figura 7.7.2 ). En la mayoría de los casos hay poca mejora en la eficiencia de extracción después de cinco o seis extracciones. Para las condiciones del Ejemplo 7.7.2 , alcanzamos una eficiencia de extracción del 99% después de cinco extracciones y necesitamos tres extracciones adicionales para obtener el incremento extra de 0.9% en la eficiencia de extracción.
Para planificar una extracción líquido-líquido necesitamos conocer la relación de distribución del soluto entre las dos fases. Un enfoque es llevar a cabo la extracción sobre una solución que contenga una cantidad conocida de soluto. Después de la extracción, aislamos la fase orgánica y dejamos que se evapore, dejando atrás el soluto. En uno de esos experimentos, se disuelven 1.235 g de un soluto con una masa molar de 117.3 g/mol en 10.00 mL de agua. Después de extraer con 5.00 mL de tolueno, se recuperan 0.889 g del soluto en la fase orgánica. a) ¿Cuál es la relación de distribución del soluto entre el agua y el tolueno? b) Si extraemos 20.00 mL de una solución acuosa que contiene el soluto usando 10.00 mL de tolueno, ¿cuál es la eficiencia de extracción? (c) ¿Cuántas extracciones necesitaremos para recuperar el 99.9% del soluto?
- Responder
-
(a) La relación de distribución del soluto entre el agua y el tolueno es
\[D=\frac{\left[S_{o r g}\right]}{\left[S_{a q}\right]}=\frac{0.889 \ \mathrm{g} \times \frac{1 \ \mathrm{mol}}{117.3 \ \mathrm{g}} \times \frac{1}{0.00500 \ \mathrm{L}}}{(1.235 \ \mathrm{g}-0.889 \ \mathrm{g}) \times \frac{1 \ \mathrm{mol}}{117.3 \ \mathrm{g}} \times \frac{1}{0.01000 \ \mathrm{L}}}=5.14 \nonumber\]
b) La fracción de soluto que queda en la fase acuosa después de una extracción es
\[\left(q_{a q}\right)_{1}=\frac{V_{a q}}{D V_{org}+V_{a q}}=\frac{20.00 \ \mathrm{mL}}{(5.14)(10.00 \ \mathrm{mL})+20.00 \ \mathrm{mL}}=0.280 \nonumber\]
La eficiencia de extracción, por lo tanto, es de 72.0%.
(c) Para extraer 99.9% del soluto se requiere
\[\left(Q_{aq}\right)_{n}=0.001=\left(\frac{20.00 \ \mathrm{mL}}{(5.14)(10.00 \ \mathrm{mL})+20.00 \ \mathrm{mL}}\right)^{n}=(0.280)^{n} \nonumber\]
\[\begin{aligned} \log (0.001) &=n \log (0.280) \\ n &=5.4 \end{aligned} \nonumber\]
un mínimo de seis extracciones.
Extracciones líquido-líquido que implican equilibrios ácido-base
Como vemos en la Ecuación\ ref {7.1}, en una simple extracción líquido-líquido la relación de distribución y el coeficiente de partición son idénticos. Como resultado, la relación de distribución no depende de la composición de la fase acuosa o de la fase orgánica. Un cambio en el pH de la fase acuosa, por ejemplo, no afectará la eficiencia de extracción del soluto cuando K D y D tengan el mismo valor.
Si el soluto participa en una o más reacciones de equilibrio adicionales dentro de una fase, entonces la relación de distribución y el coeficiente de partición pueden no ser los mismos. Por ejemplo, la Figura 7.7.3 muestra las reacciones de equilibrio que afectan la extracción del ácido débil, HA, por una fase orgánica en la que las especies iónicas no son solubles.
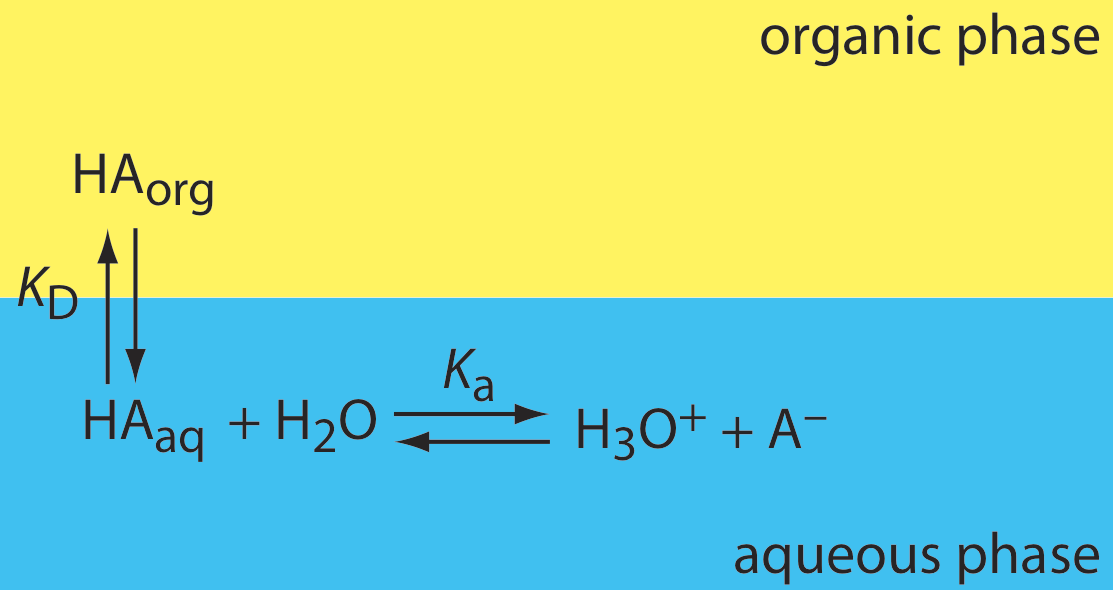
En este caso el coeficiente de partición y la relación de distribución son
\[K_{\mathrm{D}}=\frac{\left[\mathrm{HA}_{org}\right]}{\left[\mathrm{HA}_{a q}\right]} \label{7.8}\]
Debido a que la posición de un equilibrio ácido-base depende del pH, la relación de distribución, D, es dependiente del pH. Para derivar una ecuación para D que muestre esta dependencia, comenzamos con la constante de disociación ácida para HA.
Resolviendo la ecuación\ ref {7.10} para la concentración de A — en la fase acuosa
\[\left[\mathrm{A}_{a q}^{-}\right]=\frac{K_{\mathrm{a}} \times\left[\mathrm{HA}_{a q}\right]}{\left[\mathrm{H}_{3} \mathrm{O}_{a q}^{+}\right]} \nonumber\]
y sustituyendo en Ecuación\ ref {7.9} da
\[D = \frac {[\text{HA}_{org}]} {[\text{HA}_{aq}] + \frac {K_a \times [\text{HA}_{aq}]}{[\text{H}_3\text{O}_{aq}^+]}} \nonumber\]
Factorizar [HA aq] a partir del denominador, reemplazar [HA org]/[HA aq] por K D (Ecuación\ ref {7.8}), y simplificar nos deja con la siguiente relación entre la relación de distribución, D, y el pH de la solución acuosa.
Un soluto ácido, HA, tiene un K a de\(1.00 \times 10^{-5}\) y un K D entre agua y hexano de 3.00. Calcular la eficiencia de extracción si extraemos una muestra de 50.00 mL de una solución acuosa 0.025 M de HA, tamponada a un pH de 3.00, con 50.00 mL de hexano. Repita para niveles de pH de 5.00 y 7.00.
Solución
Cuando el pH es 3.00, [\(\text{H}_3\text{O}_{aq}^+\)] es\(1.0 \times 10^{-3}\) y la relación de distribución es
\[D=\frac{(3.00)\left(1.0 \times 10^{-3}\right)}{1.0 \times 10^{-3}+1.00 \times 10^{-5}}=2.97 \nonumber\]
La fracción de soluto que permanece en la fase acuosa es
\[\left(Q_{aq}\right)_{1}=\frac{50.00 \ \mathrm{mL}}{(2.97)(50.00 \ \mathrm{mL})+50.00 \ \mathrm{mL}}=0.252 \nonumber\]
La eficiencia de extracción, por lo tanto, es de casi 75%. El mismo cálculo a un pH de 5.00 da la eficiencia de extracción como 60%. A un pH de 7.00 la eficiencia de extracción es de apenas 3%.
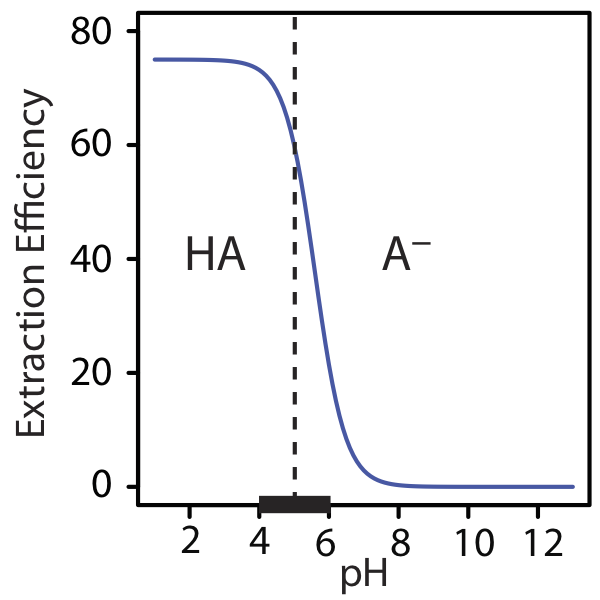
La eficiencia de extracción en el Ejemplo 7.7.3 es mayor a niveles de pH más ácidos porque el AH es la forma predominante del soluto en la fase acuosa. A un pH más básico, donde A — es la forma predominante del soluto, la eficiencia de extracción es menor. En la Figura 7.7.4 se muestra una gráfica de la eficiencia de extracción versus pH. Tenga en cuenta que la eficiencia de extracción es esencialmente independiente del pH para niveles de pH más ácidos que los pK a de HA, y que es esencialmente cero para niveles de pH más básicos que los de HA p K a. El mayor cambio en la eficiencia de extracción ocurre a niveles de pH donde tanto HA como A son especies predominantes. El diagrama de escalera para HA a lo largo del eje x de la gráfica ayuda a ilustrar este efecto.
La extracción líquido-líquido de la base débil B se rige por las siguientes reacciones de equilibrio:
\[\begin{array}{c}{\mathrm{B}(a q) \rightleftharpoons \mathrm{B}(org) \quad K_{D}=5.00} \\ {\mathrm{B}(a q)+\mathrm{H}_{2} \mathrm{O}(l)\rightleftharpoons \mathrm{OH}^{-}(a q)+\mathrm{HB}^{+}(a q) \quad K_{b}=1.0 \times 10^{-4}}\end{array} \nonumber\]
Derivar una ecuación para la relación de distribución, D, y calcular la eficiencia de extracción si 25.0 mL de una solución 0.025 M de B, tamponada a un pH de 9.00, se extrae con 50.0 mL del solvente orgánico.
- Responder
-
Debido a que la base débil existe en dos formas, solo una de las cuales extrae a la fase orgánica, el coeficiente de partición, K D, y la relación de distribución, D, no son idénticos.
\[K_{\mathrm{D}}=\frac{\left[\mathrm{B}_{org}\right]}{\left[\mathrm{B}_{aq}\right]} \nonumber\]
\[D = \frac {[\text{B}_{org}]} {[\text{B}_{aq}]} = \frac {[\text{B}_{org}]} {[\text{B}_{aq}] + [\text{HB}_{aq}^+]} \nonumber\]
Uso de la expresión K b para la base débil
\[K_{\mathrm{b}}=\frac{\left[\mathrm{OH}_{a q}^{-}\right]\left[\mathrm{HB}_{a q}^{+}\right]}{\left[\mathrm{B}_{a q}\right]} \nonumber\]
resolvemos para la concentración de HB + y sustituimos de nuevo en la ecuación para D, obteniendo
\[D = \frac {[\text{B}_{org}]} {[\text{B}_{aq}] + \frac {K_b \times [\text{B}_{aq}]} {[\text{OH}_{aq}^-]}} = \frac {[\text{B}_{org}]} {[\text{B}_{aq}]\left(1+\frac {K_b} {[\text{OH}_{aq}^+]} \right)} =\frac{K_{D}\left[\mathrm{OH}_{a q}^{-}\right]}{\left[\mathrm{OH}_{a q}^{-}\right]+K_{\mathrm{b}}} \nonumber\]
A un pH de 9.0, el [OH —] es\(1 \times 10^{-5}\) M y la relación de distribución tiene un valor de
\[D=\frac{K_{D}\left[\mathrm{OH}_{a q}^{-}\right]}{\left[\mathrm{OH}_{aq}^{-}\right]+K_{\mathrm{b}}}=\frac{(5.00)\left(1.0 \times 10^{-5}\right)}{1.0 \times 10^{-5}+1.0 \times 10^{-4}}=0.455 \nonumber\]
Después de una extracción, la fracción de B restante en la fase acuosa es
\[\left(q_{aq}\right)_{1}=\frac{25.00 \ \mathrm{mL}}{(0.455)(50.00 \ \mathrm{mL})+25.00 \ \mathrm{mL}}=0.524 \nonumber\]
La eficiencia de extracción, por lo tanto, es de 47.6%. A un pH de 9, la mayor parte de la base débil está presente como HB +, lo que explica por qué la eficiencia general de extracción es tan pobre.
Extracción líquido-líquido de un complejo metal-ligando
Una aplicación importante de una extracción líquido-líquido es la extracción selectiva de iones metálicos usando un ligando orgánico. Desafortunadamente, muchos ligandos orgánicos no son muy solubles en agua o sufren reacciones de hidrólisis u oxidación en soluciones acuosas. Por estas razones, el ligando se añade al disolvente orgánico en lugar de a la fase acuosa. La Figura 7.7.5 muestra las reacciones de equilibrio relevantes (y constantes de equilibrio) para la extracción de Mn+ por el ligando HL, incluyendo la extracción del ligando en la fase acuosa (KD, HL), la reacción de disociación ácida del ligando ( K a), la formación del complejo metal-ligando (\(\beta_n\)) y la extracción del complejo a la fase orgánica (K D, c).
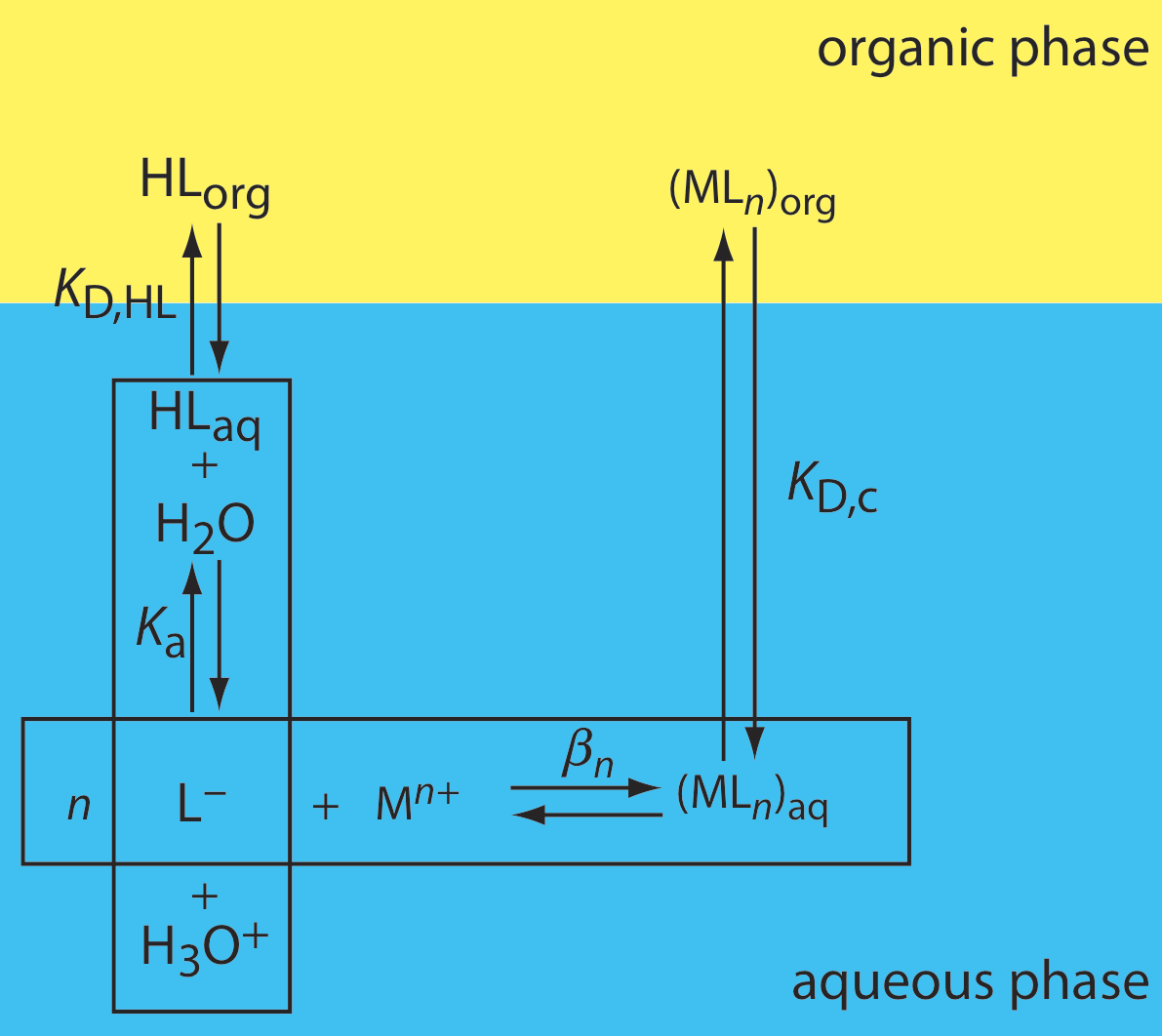
Si la concentración del ligando es mucho mayor que la concentración del ion metálico, entonces la relación de distribución es
donde C HL es la concentración inicial del ligando en la fase orgánica. Como se muestra en el Ejemplo 7.7.4 , la eficiencia de extracción para iones metálicos muestra una marcada dependencia del pH.
Una extracción líquido-líquido del ión metálico divalente, M 2+, utiliza el esquema descrito en la Figura 7.7.5 . Los coeficientes de partición para el ligando, K D, HL, y para el complejo metal-ligando, K D, c, son\(1.0 \times 10^4\) y\(7.0 \times 10^4\), respectivamente. La constante de disociación ácida del ligando, K a, es\(5.0 \times 10^{-5}\), y la constante de formación para el complejo metal-ligando,\(\beta_2\), es\(2.5 \times 10^{16}\). ¿Cuál es la eficiencia de extracción si extraemos 100.0 mL de una solución acuosa\(1.0 \times 10^{-6}\) M de M 2+, tamponada a un pH de 1.00, con 10.00 mL de un solvente orgánico que es 0.1 mM en el agente quelante? Repetir el cálculo a un pH de 3.00.
Solución
Cuando el pH es 1.00 la relación de distribución es
\[D=\frac{\left(2.5 \times 10^{16}\right)\left(7.0 \times 10^{4}\right)\left(5.0 \times 10^{-5}\right)^{2}\left(1.0 \times 10^{-4}\right)^{2}}{\left(1.0 \times 10^{4}\right)^{2}(0.10)^{2}+\left(2.5 \times 10^{16}\right)\left(5.0 \times 10^{-5}\right)^{2}\left(1.0 \times 10^{-4}\right)^{2}} \nonumber\]
o una D de 0.0438. La fracción de ion metálico que permanece en la fase acuosa es
\[\left(Q_{aq}\right)_{1}=\frac{100.0 \ \mathrm{mL}}{(0.0438)(10.00 \ \mathrm{mL})+100.0 \ \mathrm{mL}}=0.996 \nonumber\]
A un pH de 1.00, extraemos solo 0.40% del metal en la fase orgánica. Cambiando el pH a 3.00, sin embargo, aumenta la eficiencia de extracción a 97.8%. La Figura 7.7.6 muestra cómo el pH de la fase acuosa afecta la eficiencia de extracción para M 2+.
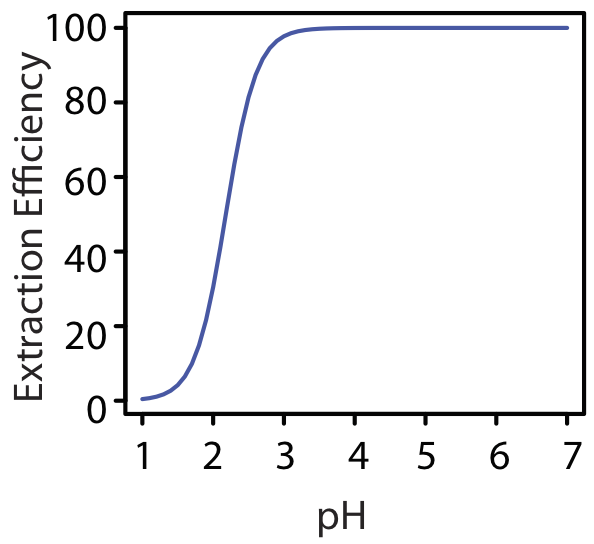
Una ventaja de usar un ligando para extraer un ion metálico es el alto grado de selectividad que aporta a una extracción líquido-líquido. Como se ve en la Figura 7.7.6 , la eficiencia de extracción de un ión metálico divalente aumenta de aproximadamente 0% a 100% en un rango de 2 unidades de pH. Debido a que la capacidad de un ligando para formar un complejo metal-ligando varía sustancialmente de ión metálico a ion metálico, es posible una selectividad significativa si controlamos cuidadosamente el pH. El Cuadro 7.7.1 muestra el pH mínimo para extraer 99% de un ión metálico de una solución acuosa usando un volumen igual de ditizona 4 mM en CCl 4.
| ión metálico | pH mínimo |
|---|---|
| Hg 2 + | —8.7 |
| Ag + | —1.7 |
| Cu 2+ | —0.8 |
| Bi 3 + | 0.9 |
| Zn 2+ | 2.3 |
| Cd 2+ | 3.6 |
| Co 2+ | 3.6 |
| Pb 2+ | 4.1 |
| Ni 2+ | 6.0 |
| Tl + | 8.7 |
Usando la Tabla 7.7.1 , explique cómo podemos separar los iones metálicos en una mezcla acuosa de Cu 2 +, Cd 2+ y Ni 2 + extrayendo con un volumen igual de ditizona en CCl 4.
Solución
De la Tabla 7.7.1 , es posible una separación cuantitativa de Cu 2 + de Cd 2 + y de Ni 2 + si acidificamos la fase acuosa a un pH inferior a 1. Este pH es mayor que el pH mínimo para extraer Cu 2 + y significativamente menor que el pH mínimo para extraer ya sea Cd 2 + o Ni 2 +. Una vez completada la extracción de Cu 2 +, cambiamos el pH de la fase acuosa a 4.0, lo que nos permite extraer Cd 2 + dejando Ni 2 + en la fase acuosa.