
7.6: Clasificación de las técnicas de separación
- Page ID
- 75941

Podemos separar un analito y un interferente si hay una diferencia significativa en al menos una de sus propiedades químicas o físicas. Table 7.6.1 proporciona una lista parcial de técnicas de separación, organizadas por la propiedad química o física que afecta a la separación.
| base de separación | técnica (s) de separación |
|---|---|
| tamaño | filtración; dialaísis; cromatografía de exclusión por tamaño |
| masa o densidad | centrifugación |
| formación de complejos | enmascaramiento |
| cambio en el estado físico | destilación; sublimación; recristalización |
| cambio en el estado químico | precipitación; electrodeposición; volatilización |
| partición entre fases | extracción; cromatografía |
Separaciones basadas en el tamaño
El tamaño es la propiedad física más simple que podemos explotar en una separación. Para lograr la separación utilizamos un medio poroso a través del cual solo puede pasar el analito o el interferente. Los ejemplos de separaciones basadas en tamaño incluyen filtración, diálisis y exclusión por tamaño.
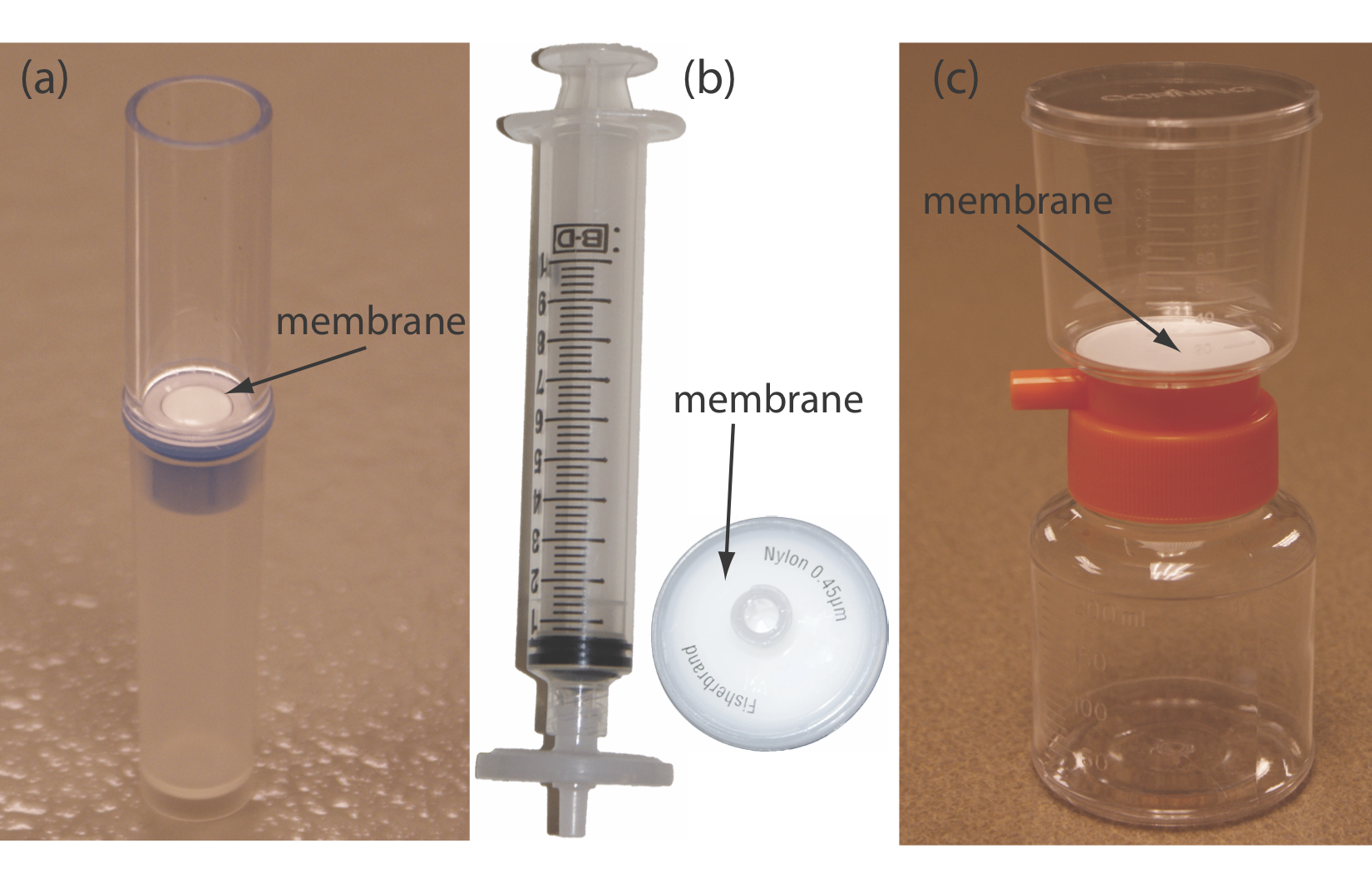
En una filtración separamos un interferente particulado de los analitos solubles usando un filtro con un tamaño de poro que retendrá el interferente. La solución que pasa a través del filtro se llama filtrado, y el material retenido por el filtro es el retenido. La filtración por gravedad y la filtración por succión utilizando papel de filtro son técnicas con las que ya deberías estar familiarizado. Un filtro de membrana es el método de elección para partículas que son demasiado pequeñas para ser retenidas por papel de filtro. La Figura 7.6.1 proporciona información sobre tres tipos de filtros de membrana. Para aplicaciones de filtración por gravedad y filtración por succión en métodos gravimétricos de análisis, ver Capítulo 8.
La diálisis es otro ejemplo de una técnica de separación en la que se utiliza el tamaño para separar el analito y el interferente. Una membrana de diálisis generalmente se fabrica con celulosa y se forma en tubos, bolsas o casetes. La Figura 7.6.2 muestra un ejemplo de un casete de diálisis disponible comercialmente. La muestra se inyecta en la membrana de diálisis, la cual se sella herméticamente mediante una junta, y la unidad se coloca en un recipiente lleno de una solución con una composición diferente a la muestra. Si hay una diferencia en la concentración de una especie en los dos lados de la membrana, el gradiente de concentración resultante proporciona una fuerza impulsora para su difusión a través de la membrana. Mientras que las especies pequeñas pasan libremente a través de la membrana, las especies más grandes no pueden pasar. La diálisis se usa frecuentemente para purificar proteínas, hormonas y enzimas. Durante la diálisis renal, los productos de desecho metabólicos, como la urea, el ácido úrico y la creatinina, se eliminan de la sangre haciéndola pasar por una membrana de diálisis.

La cromatografía de exclusión por tamaño es un tercer ejemplo de una técnica de separación que utiliza el tamaño como medio para efectuar una separación. En esta técnica se empaqueta una columna con pequeñas perlas poliméricas porosas de aproximadamente 10 μm de dextrina reticulada o poliacrilamida. El tamaño de poro de las partículas está controlado por el grado de reticulación, con más reticulación produciendo tamaños de poro más pequeños. La muestra se coloca en una corriente de disolvente que se bombea a través de la columna a un caudal fijo. Aquellas especies demasiado grandes para entrar en los poros pasan a través de la columna a la misma velocidad que el disolvente. Las especies que entran en los poros tardan más en pasar por la columna, y las especies más pequeñas requieren más tiempo para pasar por la columna. La cromatografía de exclusión por tamaño es ampliamente utilizada en el análisis de polímeros, y en bioquímica, donde se usa para la separación de proteínas. Un tratamiento más detallado de la cromatografía de exclusión por tamaño, que también se denomina cromatografía de permeación en gel, se encuentra en el Capítulo 12.
Separaciones Basadas en Masa o Densidad
Si el analito y el interferente tienen diferentes masas o densidades, entonces puede ser posible una separación mediante centrifugación. La muestra se coloca en un tubo de centrífuga y se hace girar a una alta velocidad angular, medida en revoluciones por minuto (rpm). Los componentes de la muestra experimentan una fuerza centrífuga que los tira hacia el fondo del tubo de centrífuga. Aquellas especies que experimentan la mayor fuerza centrífuga tienen la velocidad de sedimentación más rápida y son las primeras en llegar al fondo del tubo de centrífuga. Si dos especies tienen la misma densidad, su separación se basa en una diferencia de masa, teniendo las especies más pesadas la mayor tasa de sedimentación. Si las especies son de igual masa, entonces la especie con mayor densidad tiene la mayor tasa de sedimentación.
La centrifugación es una técnica de separación importante en bioquímica. Table 7.6.2 , por ejemplo, enumera las condiciones para separar componentes celulares seleccionados. Podemos separar los lisosomas de otros componentes celulares mediante varias centrifugaciones diferenciales, en las que dividimos la muestra en un residuo sólido y una solución sobrenadante. Después de destruir las células, la solución se centrifuga durante 20 minutos a\(15000 \times g\) (una fuerza centrífuga que es 15 000 veces la fuerza gravitacional de la tierra), dejando un residuo sólido de membranas celulares y mitocondrias. El sobrenadante, que contiene los lisosomas, se aísla decantándolo del residuo y luego se centrifuga durante 30 minutos a\(30000 \times g\), dejando un residuo sólido de lisosomas. La Figura 7.6.3 muestra una centrífuga típica capaz de producir las fuerzas centrífugas necesarias para las separaciones bioquímicas.
| componentes | fuerza centrífuga (\(\times g\)) | tiempo (min) |
|---|---|---|
| células eucariotas | \ (\ veces g\)) ">1000 | 5 |
| membranas celulares; núcleos | \ (\ veces g\)) ">4000 | 10 |
| mitocondrias, células bacterianas | \ (\ veces g\)) ">15000 | 20 |
| lisosomas; membranas bacterianas | \ (\ veces g\)) ">30000 | 30 |
| ribosomas | \ (\ veces g\)) ">100000 | 180 |
|
Fuente: Adaptado de Zubay, G. Bioquímica, 2a ed. Macmillan: Nueva York, 1988, p.120. |
||

Un enfoque alternativo a la centrifugación diferencial es una centrifugación en gradiente de densidad. Para preparar un gradiente de densidad de sacarosa, por ejemplo, una solución con una menor concentración de sacarosa y, por lo tanto, de menor densidad, se coloca suavemente sobre una solución con una mayor concentración de sacarosa. Repitiendo este proceso varias veces, llena el tubo de centrífuga con un gradiente de densidad multicapa. La muestra se coloca encima del gradiente de densidad y se centrifuga utilizando una fuerza mayor que\(150000 \times g\). Durante la centrifugación, cada uno de los componentes de la muestra se mueve a través del gradiente hasta alcanzar una posición donde su densidad coincide con la solución de sacarosa circundante. Cada componente se aísla como una banda separada posicionada donde su densidad es igual a la de la densidad local dentro del gradiente. La Figura 7.6.4 proporciona un ejemplo de una centrifugación típica de densidad de sacarosa para separar membranas tilacoides de plantas.

Separaciones basadas en reacciones de complejación (enmascaramiento)
Una técnica ampliamente utilizada para prevenir una interferencia es unir al interferente en un complejo fuerte y soluble que evite que interfiera en la determinación del analito. Este proceso se conoce como enmascaramiento. Como se muestra en la Tabla 7.6.3 , una amplia variedad de iones y moléculas son agentes enmascarantes útiles y, como resultado, la selectividad no suele ser un problema.
Técnicamente, el enmascaramiento no es una técnica de separación porque no separamos físicamente el analito y el interferente. Sin embargo, aislamos químicamente el interferente del analito, dando como resultado una pseudoseparación.
| agente de enmascaramiento | elementos cuyos iones están enmascarados |
|---|---|
| CN — |
Ag, Au, Cd, Co, Cu, Fe, Hg, Mn, Ni, Pd, Pt, Zn |
| SCN — |
Ag, Cd, Co, Cu, Fe, Ni, Pd, Pt, Zn |
| NH 3 |
Ag, Co, Ni, Cu, Zn |
| F — | Al, Co, Cr, Mg, Mn, Sn, Zn |
| \(\text{S}_2\text{O}_3^{2-}\) |
Au, Ce, Co, Cu, Fe, Hg, Mn, Pb, Pd, Pt, Sb, Sn, Zn |
| tartrato | Al, Ba, Bi, Ca, Ce, Co, Cr, Cu, Fe, Hg, Mn, Pb, Pd, Pt, Sb, Sn, Zn |
| oxalato |
Al, Fe, Mg, Mn |
| ácido tioglicólico |
Cu, Fe, Sn |
|
Fuente: Meites, L. Handbook of Analytical Chemistry, McGraw-Hill: Nueva York, 1963. |
|
Usando la Tabla 7.6.3 , sugerir un agente enmascarante para el análisis del aluminio en presencia de hierro.
Solución
Un agente enmascarante adecuado debe formar un complejo con el interferente, pero no con el analito. El oxalato, por ejemplo, no es un agente enmascarante adecuado porque se une tanto al como al fe. El ácido tioglicólico, por otro lado, es un agente enmascarante selectivo para Fe en presencia de Al. Otros agentes enmascarantes aceptables son cianuro (CN —) tiocianato (SCN —) y tiosulfato (\(\text{S}_2\text{O}_3^{2-}\)).
Usando el Cuadro 7.6, sugerir un agente enmascarante para el análisis de Fe en presencia de Al.
- Contestar
-
El ion fluoruro, F —, es un agente enmascarante adecuado ya que se une con Al 3 + para formar el\(\text{AlF}_6^{3-}\) complejo estable, dejando hierro en solución.
Como se muestra en el Ejemplo 7.6.2 , podemos juzgar la efectividad de un agente enmascarante considerando las constantes de equilibrio relevantes.
Mostrar que CN — es un agente enmascarante apropiado para Ni 2 + en un método donde la complejación de níquel con EDTA es una interferencia.
Solución
Las reacciones y constantes de formación relevantes son
\[\mathrm{Ni}^{2+}(a q)+\mathrm{Y}^{4-}(a q)\rightleftharpoons \mathrm{NiY}^{2-}(a q) \quad K_{1}=4.2 \times 10^{18} \nonumber\]
\[\mathrm{Ni}^{2+}(a q)+4 \mathrm{CN}^{-}(a q)\rightleftharpoons \mathrm{Ni}(\mathrm{CN})_{4}^{2-}(a q) \quad \beta_{4}=1.7 \times 10^{30} \nonumber\]
donde Y 4— es una abreviatura de EDTA. El cianuro es un agente enmascarante apropiado porque la constante de formación\(\text{Ni(CN)}_4^{2-}\) es mayor que la del complejo Ni—EDTA. De hecho, la constante de equilibrio para la reacción en la que EDTA desplaza al agente enmascarante
\[\mathrm{Ni}(\mathrm{CN})_{4}^{2-}(a q)+\mathrm{Y}^{4-}(a q) \rightleftharpoons \mathrm{NiY}^{2-}(a q)+4 \mathrm{CN}^{-}(a q) \nonumber\]
\[K=\frac{K_{1}}{\beta_{4}}=\frac{4.2 \times 10^{18}}{1.7 \times 10^{30}}=2.5 \times 10^{-12} \nonumber\]
es suficientemente pequeño que\(\text{Ni(CN)}_4^{2-}\) es relativamente inerte en presencia de EDTA.
Utilice las constantes de formación del Apéndice 12 para demostrar que 1,10-fenantrolina es un agente enmascarante adecuado para Fe 2 + en presencia de Fe 3 +. Use un diagrama de escalera para definir cualquier limitación en el uso de 1,10-fenantrolina como agente enmascarante. Consulte el Capítulo 6 para una revisión de los diagramas de escalera.
- Contestar
-
Las reacciones relevantes y constantes de equilibrio son
\[\begin{array}{ll}{\mathrm{Fe}^{2+}(a q)+3 \mathrm{phen}(a q)} & {\rightleftharpoons\mathrm{Fe}(\mathrm{phen})_{3}^{2+}(a q) \quad \beta_{3}=5 \times 10^{20}} \\ {\mathrm{Fe}^{3+}(a q)+3 \mathrm{phen}(a q)} & {\rightleftharpoons \mathrm{Fe}(\mathrm{phen})_{3}^{3+}(a q) \quad \beta_{3}=6 \times 10^{13}}\end{array} \nonumber\]
donde phen es una abreviatura de 1,10-fenantrolina. Porque\(\beta_3\) es más grande para el complejo con Fe 2 + que para el complejo con Fe 3 + ,1,10-fenantrolina se unirá a Fe 2 + antes de que se una a Fe 3 +. Un diagrama de escalera para este sistema (como se muestra a continuación) sugiere que un equilibrio p (phen) entre 5.6 y 5.9 complejará completamente Fe 2 + sin ninguna formación significativa del\(\text{Fe(phen)}_3^{3+}\) complejo. Agregar una cantidad estequiométricamente equivalente de 1,10-fenantrolina a una solución de Fe 2 + es suficiente para enmascarar Fe 2 + en presencia de Fe 3 +. Sin embargo, un gran exceso de 1,10-fenantrolina disminuye p (phen) y permite la formación de ambos complejos metal-ligando.

Separaciones basadas en un cambio de Estado
Debido a que un analito y su interferente suelen estar en la misma fase, podemos lograr una separación si uno de ellos sufre un cambio en su estado físico o su estado químico.
Cambios en el Estado Físico
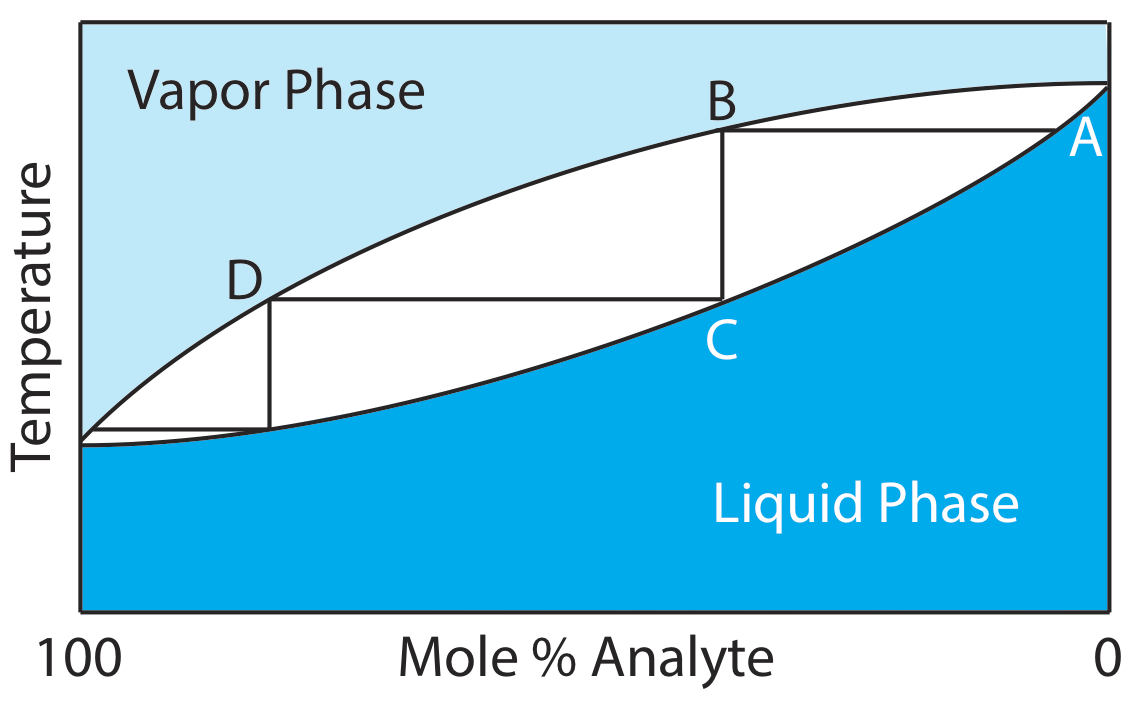
Cuando el analito y el interferente son líquidos miscibles, la separación por destilación es posible si sus puntos de ebullición son significativamente diferentes. La Figura 7.6.5 muestra el progreso de una destilación como una gráfica de temperatura frente a la composición de la fase vapor y la fase líquida de la mezcla. La mezcla líquida inicial (punto A), contiene más interferente que analito. Cuando esta solución se lleva a su punto de ebullición, la fase vapor en equilibrio con la fase líquida se enriquece en analito (punto B). La línea horizontal que conecta los puntos A y B representa este equilibrio de vaporización. Condensar la fase vapor en el punto B, al bajar la temperatura, crea una nueva fase líquida con una composición idéntica a la de la fase vapor (punto C). La línea vertical que conecta los puntos B y C representa este equilibrio de condensación. La fase líquida en el punto C tiene un punto de ebullición menor que la mezcla original, y se encuentra en equilibrio con la fase vapor en el punto D. Este proceso de vaporización y condensación repetidas separa gradualmente el analito y el interferente.
Dos configuraciones experimentales para destilaciones se muestran en la Figura 7.6.6 . El aparato de destilación simple que se muestra en la Figura 7.6.6 a es útil solo para separar un analito volátil (o interferente) de un interferente no volátil (o analito), o para separar un analito y un interferente cuyos puntos de ebullición difieren en más de 150 oC. la separación se logra utilizando el aparato de destilación fraccionada de la Figura 7.6.6 b. El empaquetamiento de la columna de fraccionamiento con un material de alta área superficial, como una esponja de acero o perlas de vidrio, brinda más oportunidad para el proceso repetido de vaporización y condensación necesario para efectuar una separación completa.

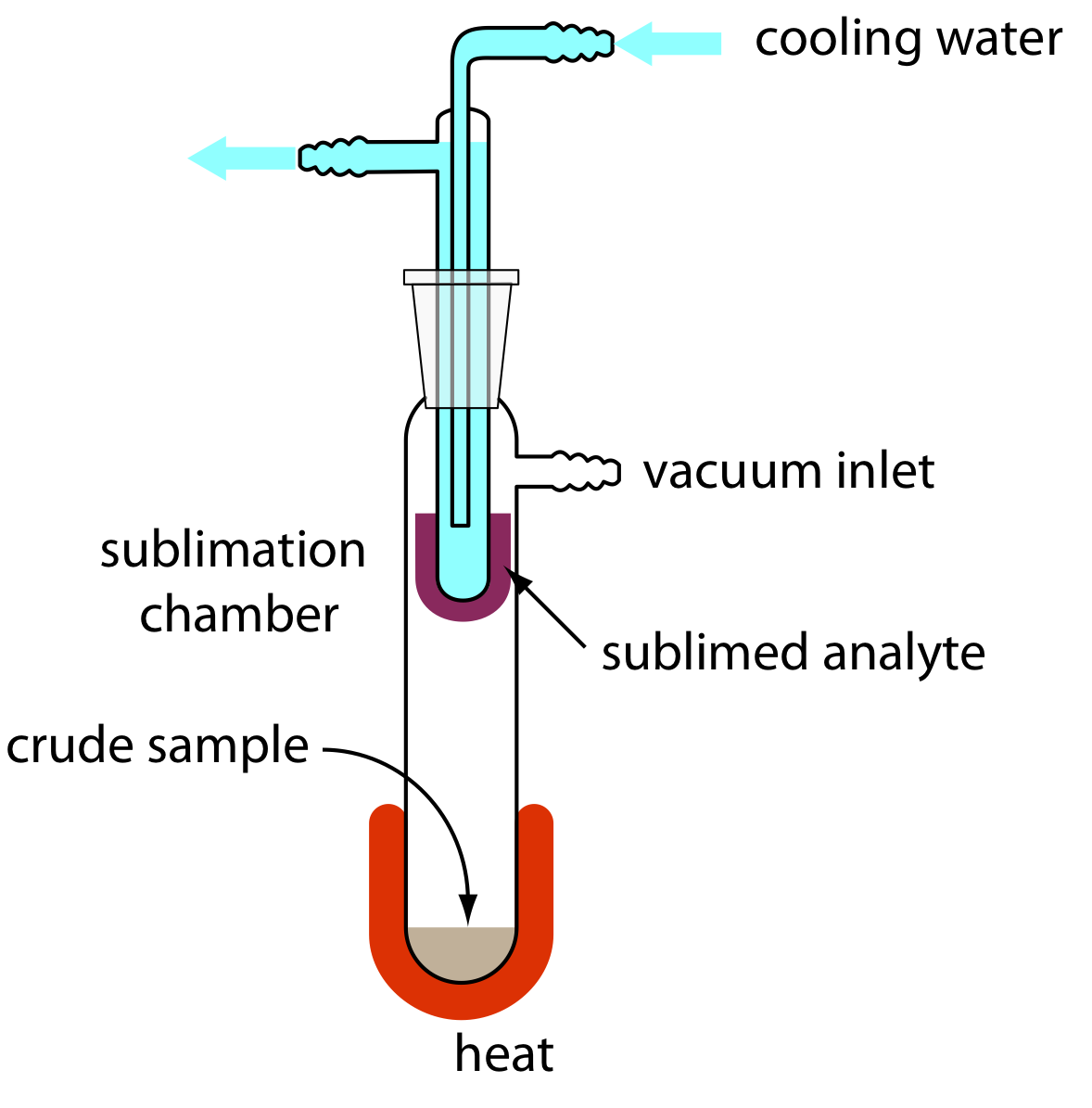
Cuando la muestra es un sólido, la sublimación puede proporcionar una separación útil del analito y el interferente. La muestra se calienta a una temperatura y presión por debajo del punto triple del analito, permitiendo que se vaporice sin pasar por un estado líquido. Condensar el vapor recupera el analito purificado (Figura 7.6.7 ). Un ejemplo analítico útil de sublimación es el aislamiento de aminoácidos de conchas de moluscos fósiles y sedimentos de aguas profundas [Glavin, D. P.; Bada, J. L. Anal. Chem. 1998, 70, 3119—3122].
La recristalización es otro método para purificar un sólido. Se elige un disolvente en el que la solubilidad del analito sea significativa cuando el disolvente está caliente y mínima cuando el disolvente está frío. Los interferentes deben ser menos solubles en el disolvente caliente que el analito o estar presentes en cantidades mucho menores. Después de calentar una porción del disolvente en un matraz Erlenmeyer, se agregan pequeñas cantidades de muestra hasta que la muestra no disuelta sea visible. Se agrega disolvente caliente adicional hasta que la muestra se redisuelve, o hasta que solo quedan impurezas insolubles. Este proceso de adición de muestra y disolvente se repite hasta que se agrega toda la muestra al matraz Erlenmeyer. Cualquier impureza insoluble se elimina filtrando la solución caliente. La solución se deja enfriar lentamente, lo que promueve el crecimiento de cristales grandes y puros, y luego se enfría en un baño de hielo para minimizar las pérdidas de solubilidad. La muestra purificada se aísla por filtración y se enjuaga para eliminar cualquier impureza soluble. Finalmente, la muestra se seca para eliminar cualquier resto de trazas del disolvente. La purificación adicional, si es necesario, se realiza mediante recristalizaciones adicionales.
Cambios en el Estado Químico
La destilación, sublimación y recristalización utilizan un cambio en el estado físico para efectuar una separación. La reactividad química también es una herramienta útil para separar analitos e interferentes. Por ejemplo, podemos separar SiO 2 de una muestra haciéndola reaccionar con HF para formar SiF 4. Debido a que el SiF 4 es volátil, es fácil de eliminar por evaporación. Si queremos recoger el producto volátil de la reacción, entonces es posible una destilación. Por ejemplo, podemos aislar el\(\text{NH}_4^+\) en una muestra haciendo básica la solución y convirtiéndola en NH 3. Luego se elimina el amoníaco por destilación. El cuadro 7.6.4 proporciona ejemplos adicionales de este enfoque para aislar iones inorgánicos.
| analito | tratamiento | especies aisladas |
|---|---|---|
| \(\text{CO}_3^{2-}\) | \(\mathrm{CO}_{3}^{2-}(a q)+2 \mathrm{H}_{3} \mathrm{O}^{+}(a q) \rightarrow \mathrm{CO}_{2}(g)+3 \mathrm{H}_{2} \mathrm{O}(l)\) | CO 2 |
| \(\text{NH}_4^+\) | \(\mathrm{NH}_{4}^{+}(a q)+\mathrm{OH}^{-}(a q) \rightarrow \mathrm{NH}_{3}(a q)+\mathrm{H}_{2} \mathrm{O}(l)\) | NH 3 |
| \(\text{SO}_3^-\) | \(\mathrm{SO}_{3}^{2-}(a q)+2 \mathrm{H}_{3} \mathrm{O}^{+}(a q) \rightarrow \mathrm{SO}_{2}(g)+3 \mathrm{H}_{2} \mathrm{O}(l)\) | SO 2 |
| S 2— | \(\text{S}^{2-}(a q)+2 \text{H}_{3} \mathrm{O}^{+}(a q) \rightarrow \text{H}_{2} \text{S}(g)+2 \text{H}_{2} \text{O}(l)\) | H 2 S |
Otra reacción para separar analitos e interferentes es la precipitación. Dos ejemplos importantes del uso de una reacción de precipitación en una separación son la solubilidad dependiente del pH de los óxidos e hidróxidos metálicos, y la solubilidad dependiente del pH de los sulfuros metálicos.
Las separaciones basadas en la solubilidad dependiente del pH de óxidos e hidróxidos suelen utilizar un ácido fuerte, una base fuerte o un tampón NH 3/NH 4 Cl para ajustar el pH. La mayoría de los óxidos e hidróxidos metálicos son solubles en HNO 3 concentrado en caliente, aunque algunos óxidos, como WO 3, SiO 2 y SnO 2 permanecen insolubles incluso en estas duras condiciones. Para determinar la cantidad de Cu en latón, por ejemplo, podemos evitar una interferencia del Sn disolviendo la muestra con un ácido fuerte y filtrando para eliminar el residuo sólido de SnO 2.
La mayoría de los metales forman un precipitado de hidróxido en presencia de NaOH concentrado. Aquellos metales que forman hidróxidos anfóteros, sin embargo, no precipitan porque reaccionan para formar hidroxicomplejos de orden superior. Por ejemplo, Zn 2 + y Al 3 + no precipitan en NaOH concentrado porque forman los complejos solubles\(\text{Zn(OH)}_3^-\) y\(\text{Al(OH)}_4^-\). La solubilidad de Al 3 + en NaOH concentrado nos permite aislar aluminio de muestras impuras de bauxita, un mineral de Al 2 O 3. Después de triturar el mineral, lo colocamos en una solución de NaOH concentrado, disolviendo el Al 2 O 3 y formando\(\text{Al(OH)}_4^-\). Otros óxidos en el mineral, como Fe 2 O 3 y SiO 2, permanecen insolubles. Después de filtrar, recuperamos el aluminio como un precipitado de Al (OH) 3 neutralizando parte del OH — con ácido.
El pH de un tampón NH 3/NH 4 Cl (p K a = 9.26) es suficiente para precipitar la mayoría de los metales como hidróxido. Las tierras alcalinas y los metales alcalinos, sin embargo, no precipitan a este pH. Además, los iones metálicos que forman complejos solubles con NH 3, como Cu 2 +, Zn 2+, Ni 2 + y Co 2 + tampoco precipitan bajo estas condiciones.
El uso de S 2— como reactivo precipitante es uno de los primeros ejemplos de una técnica de separación. En el texto de Fresenius de 1881 A System of Instruction in Quantitative Chemical Analysis, el sulfuro se utiliza frecuentemente para separar iones metálicos del resto de la matriz de la muestra [Fresenius. C. R. A System of Instruction in Quantitative Chemical Analysis, John Wiley and Sons: New York, 1881]. El sulfuro es un reactivo útil para separar iones metálicos por dos razones: (1) la mayoría de los iones metálicos, excepto los alcalinotérreos y los metales alcalinos, forman sulfuros insolubles; y (2) estos sulfuros metálicos muestran una variación sustancial en la solubilidad. Debido a que la concentración de S2— es dependiente del pH, podemos controlar qué iones metálicos precipitan ajustando el pH. Por ejemplo, en el procedimiento gravimétrico de Fresenius para la determinación de Ni en muestras de mineral (ver Figura 1.1.1 para un diagrama esquemático de este procedimiento), se utiliza sulfuro tres veces para separar Co 2 + y Ni 2 + de Cu 2 + y, en menor medida, de Pb 2 +.
Separaciones Basadas en una Partición Entre Fases
El grupo más importante de técnicas de separación utiliza una partición selectiva del analito o interferente entre dos fases inmiscibles. Si ponemos en contacto una fase que contiene el soluto, S, con una segunda fase, el soluto se repartirá entre las dos fases, como lo demuestra la siguiente reacción de equilibrio.
\[S_{\text { phase } 1} \rightleftharpoons S_{\text { phase } 2} \label{7.1}\]
La constante de equilibrio para la reacción\ ref {7.1}
\[K_{\mathrm{D}}=\frac{\left[S_{\mathrm{phase} \ 2}\right]}{\left[S_{\mathrm{phase} \ 1}\right]} \nonumber\]
se denomina constante de distribución o coeficiente de partición. Si K D es suficientemente grande, entonces el soluto pasa de la fase 1 a la fase 2. El soluto permanecerá en la fase 1 si el coeficiente de partición es suficientemente pequeño. Cuando ponemos en contacto una fase que contiene dos solutos con una segunda fase, es posible una separación de los solutos si K D es favorable para solo uno de los solutos. Los estados físicos de las fases se identifican cuando se describe el proceso de separación, con la fase que contiene la muestra listada primero. Por ejemplo, si la muestra está en fase líquida y la segunda fase es un sólido, entonces la separación implica el reparto líquido-sólido.
Extracción entre dos fases
Llamamos al proceso de mover una especie de una fase a otra fase una extracción. Las extracciones simples son particularmente útiles para separaciones donde solo un componente tiene un coeficiente de partición favorable. Varias técnicas de separación importantes se basan en una extracción simple, incluyendo las extracciones líquido-líquido, líquido-sólido, sólido-líquido y gas—sólido.
Extracciones Líquido-Líquido
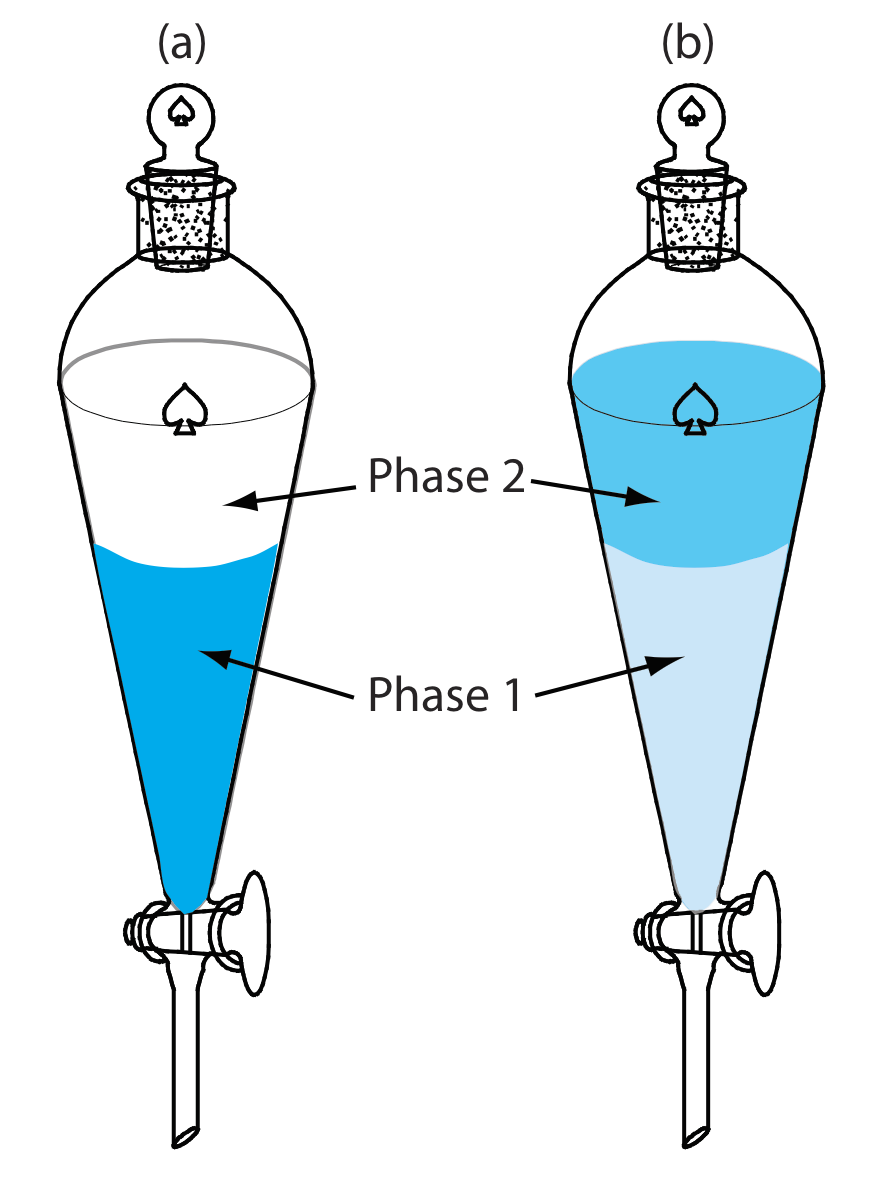
Una extracción líquido-líquido generalmente se realiza usando un embudo separador (Figura 7.6.8 ). Después de colocar los dos líquidos en el embudo separador, agitamos el embudo para aumentar la superficie entre las fases. Cuando se completa la extracción, permitimos que los líquidos se separen. La llave de paso en la parte inferior del embudo separador nos permite retirar las dos fases.
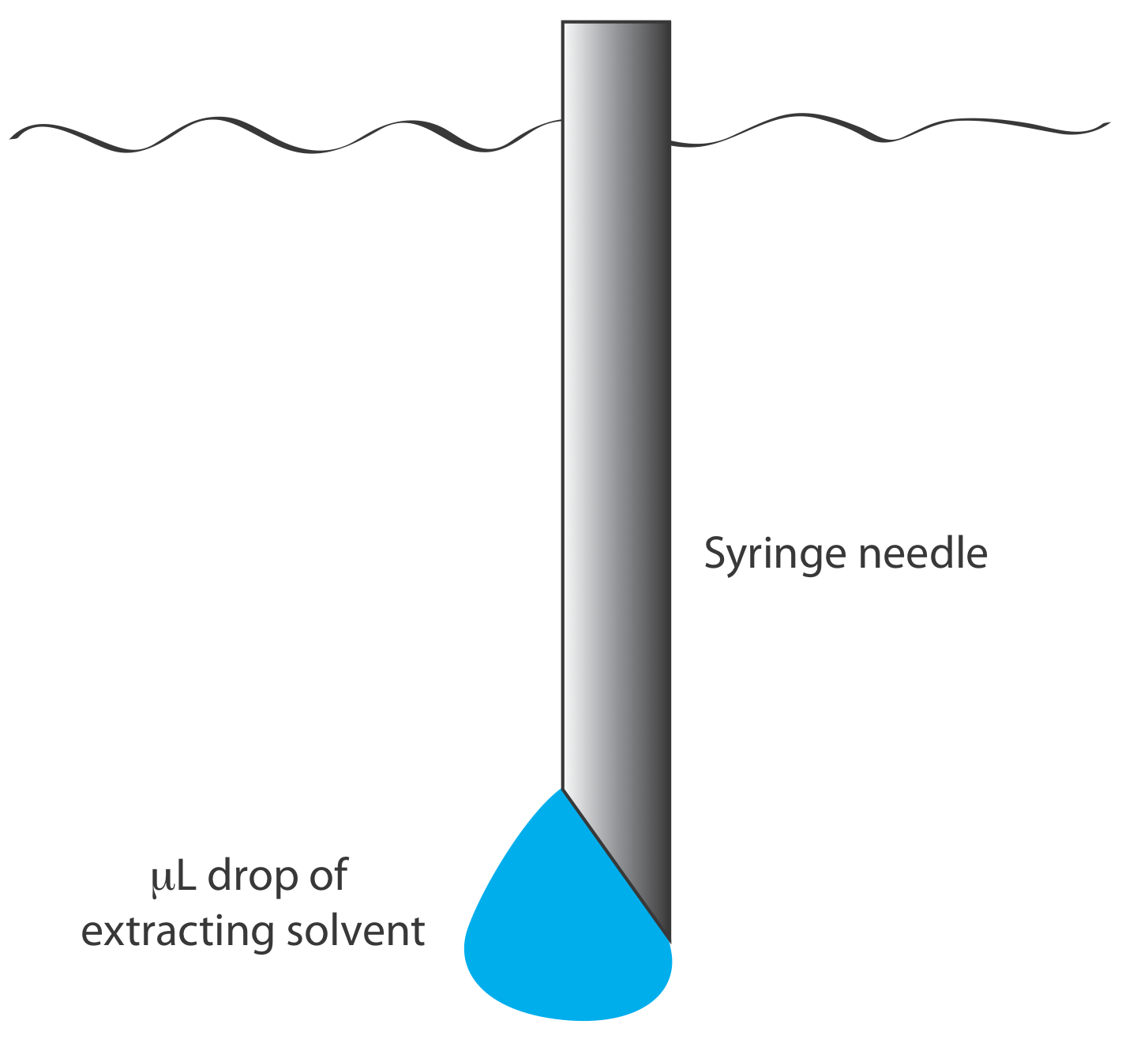
También podemos llevar a cabo una extracción líquido-líquido sin embudo separador agregando el solvente de extracción al recipiente de la muestra. Los pesticidas en el agua, por ejemplo, se conservan en el campo extrayéndolos en un pequeño volumen de hexano. También se ha descrito una microextracción líquido-líquido, en la que la fase de extracción es una gota de 1 µL suspendida de una microjeringa (Figura 7.6.9 ), también se ha descrito [Jeannot, M. A.; Cantwell, F. F. Anal. Chem. 1997, 69, 235—239]. Por su importancia, una discusión más profunda sobre las extracciones líquido-líquido se encuentra en el Capítulo 7.7.
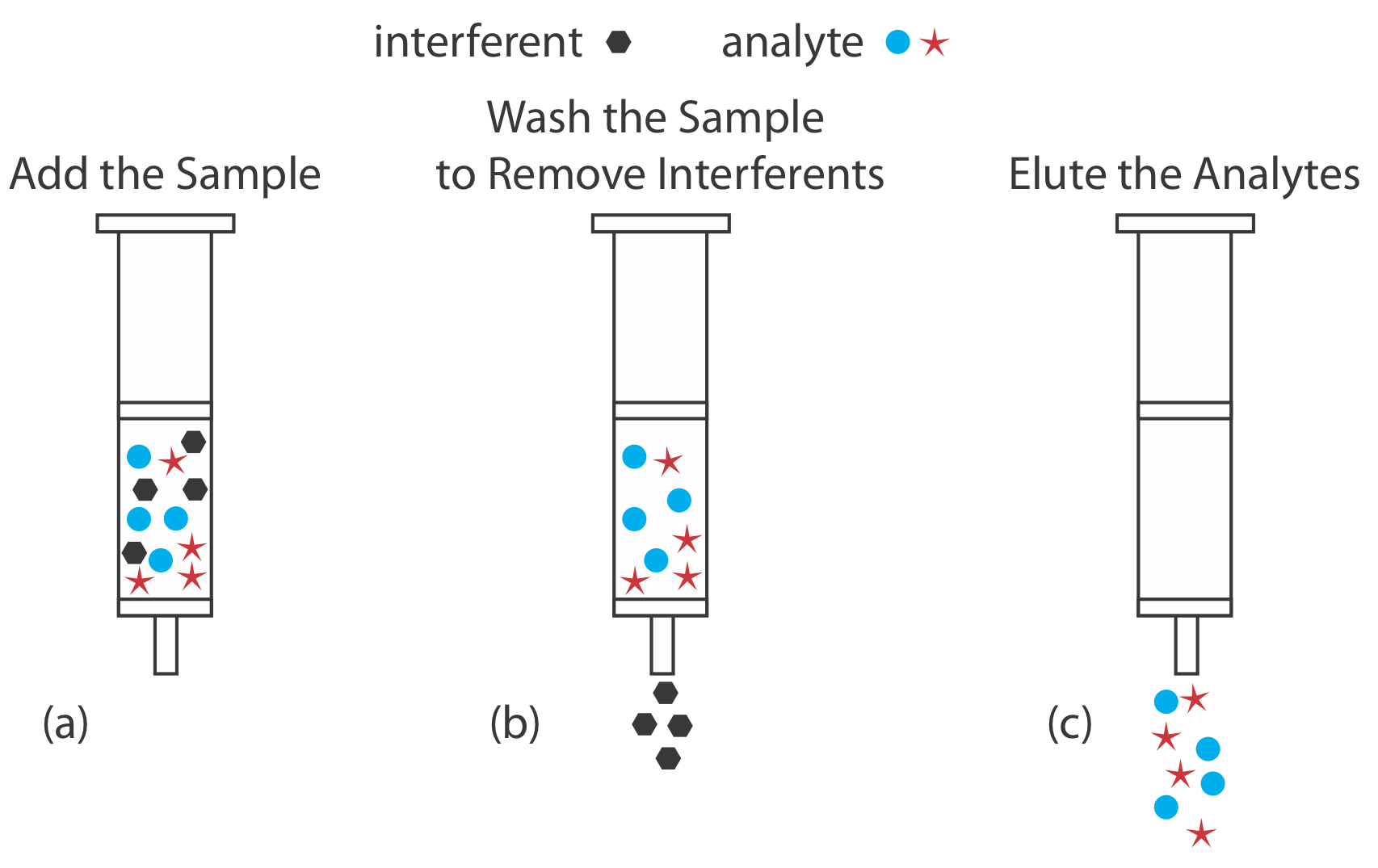
Extracciones en Fase Sólida
En una extracción en fase sólida de una muestra líquida, pasamos la muestra a través de un cartucho que contiene un adsorbente sólido, varios ejemplos de los cuales se muestran en la Figura 7.6.10 . La elección del adsorbente está determinada por las especies que deseamos separar. El cuadro 7.6.5 proporciona varios ejemplos representativos de adsorbentes sólidos y sus aplicaciones.

| absorbente | estructura | propiedades y usos |
|---|---|---|
| sílice | 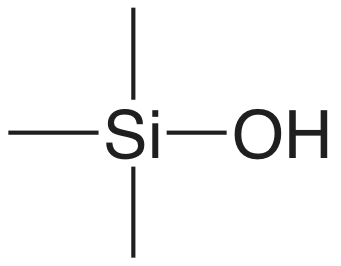 |
|
| aminopropilo | 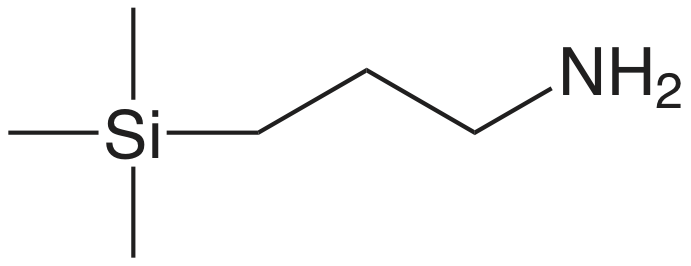 |
|
| cianopropilo | 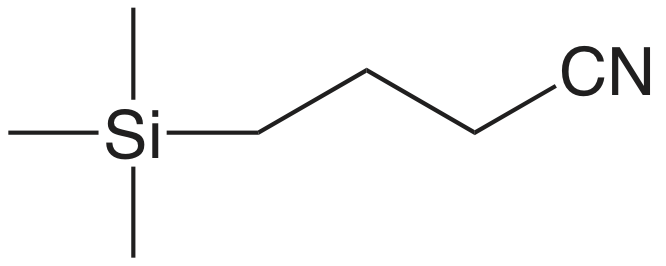 |
|
| diol | 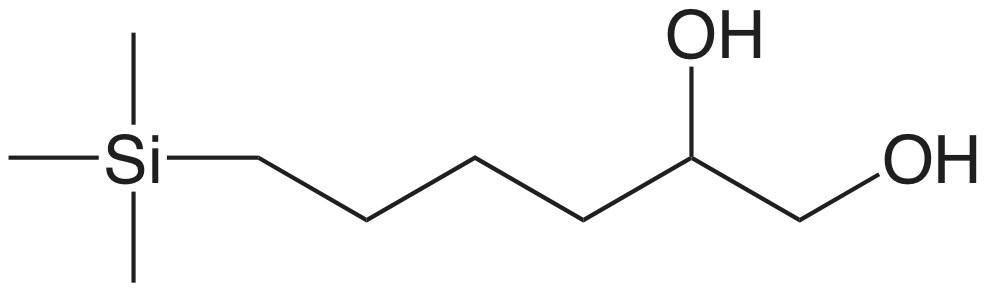 |
|
| octadecilo (C—18) | —C 18 H 37 |
|
| octilo (C—8) | —C 8 H 17 |
|
A modo de ejemplo, examinemos un procedimiento para aislar los sedantes secobarbital y fenobarbital de muestras de suero usando un adsorbente sólido C-18 [Alltech Associates Extract-Clean SPE SPE SPE Sample Preparation Guide, Bulletin 83]. Antes de agregar la muestra, el cartucho de fase sólida se enjuaga con 6 mL cada uno de metanol y agua. A continuación, se extrae una muestra de 500 μL de suero a través del cartucho, con los sedantes e interferentes de matriz retenidos después de una extracción líquido-sólido (Figura 7.6.11 a). Lavar el cartucho con agua destilada elimina cualquier interferente (Figura 7.6.11 b). Finalmente, eluimos los sedantes usando 500 μL de acetona (Figura 7.6.11 c). En comparación con una extracción líquido-líquido, una extracción en fase sólida tiene la ventaja de ser más fácil, rápida y requiere menos solvente.
Extracciones continuas
Una extracción es posible incluso si el analito tiene un coeficiente de partición desfavorable, siempre que los demás componentes de la muestra tengan coeficientes de partición significativamente menores. Debido a que el coeficiente de reparto del analito es desfavorable, una sola extracción no recuperará todo el analito. En su lugar pasamos continuamente la fase de extracción a través de la muestra hasta lograr una extracción cuantitativa.
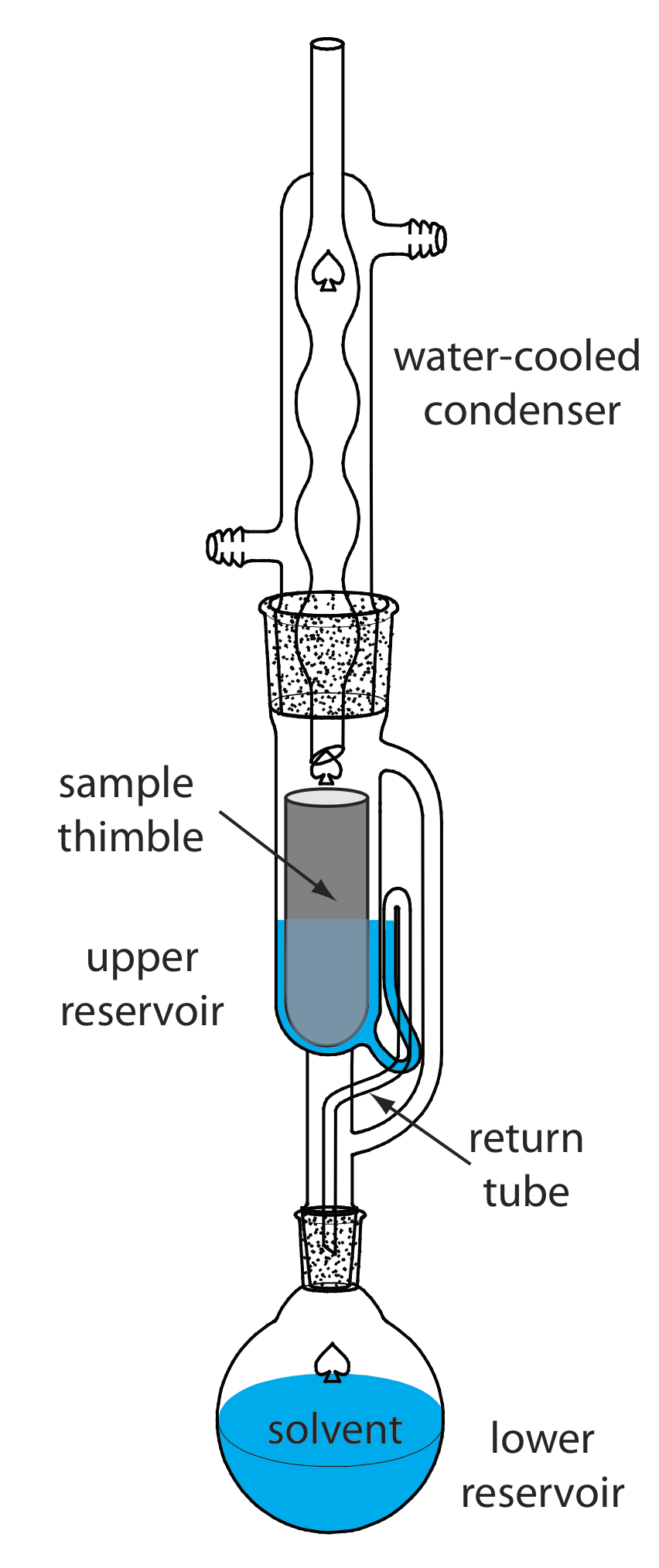
Se realiza una extracción continua de una muestra sólida utilizando un extractor Soxhlet (Figura 7.6.12 ). El disolvente de extracción se coloca en el depósito inferior y se calienta a su punto de ebullición. El solvente en la fase vapor se mueve hacia arriba a través del tubo en el extremo derecho del aparato, llegando al condensador donde se condensa de nuevo al estado líquido. Luego el disolvente pasa a través de la muestra, la cual se mantiene en un dedal de filtro de celulosa porosa, recogiéndose en el reservorio superior. Cuando el solvente en el reservorio superior alcanza la curva superior del tubo de retorno, el solvente y el analito extraído se sifonan de nuevo al reservorio inferior. Con el tiempo aumenta la concentración del analito en el reservorio inferior.
Las extracciones asistidas por microondas han reemplazado a las extracciones Soxhlet en algunas aplicaciones [Renoe, B. W. Am. Lab Agosto 1994, 34—40]. El proceso es el mismo que el descrito anteriormente para una digestión por microondas. Después de colocar la muestra y el disolvente en un recipiente de digestión sellado, se utiliza un horno de microondas para calentar la mezcla. El uso de un recipiente de digestión sellado permite que la extracción se realice a una temperatura y presión más altas, reduciendo la cantidad de tiempo necesario para una extracción cuantitativa. En una extracción Soxhlet la temperatura está limitada por el punto de ebullición del disolvente a presión atmosférica. Cuando la acetona es el disolvente, por ejemplo, una extracción Soxhlet se limita a 56 o C, pero una extracción por microondas puede alcanzar los 150 o C.
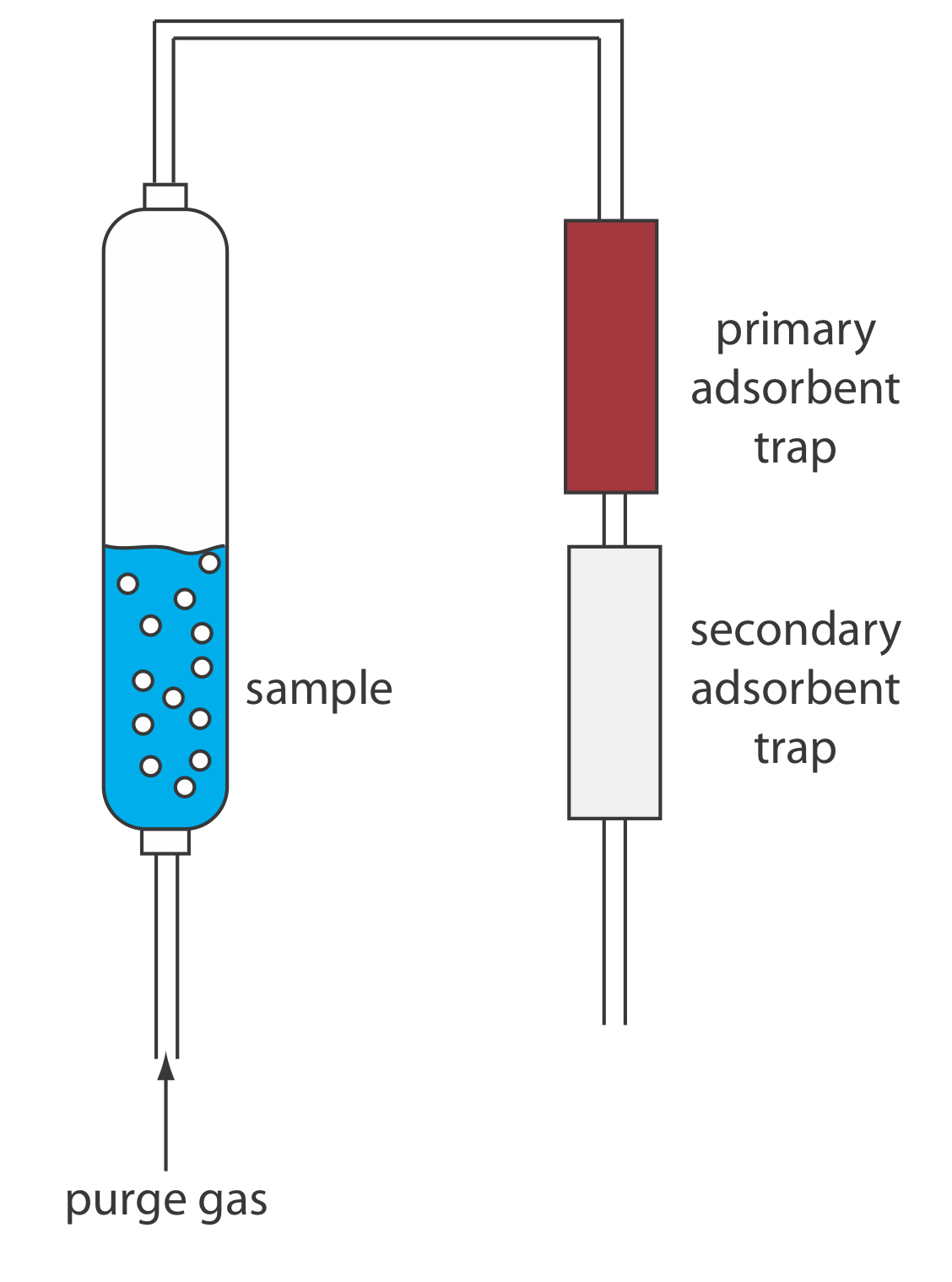
Otras dos extracciones continuas merecen mención. Los compuestos orgánicos volátiles (COV) se pueden eliminar cuantitativamente de una muestra líquida mediante una extracción líquido-gas. Como se muestra en la Figura 7.6.13 , un gas inerte de purga, como He, se pasa a través de la muestra. El gas de purga elimina los COV, los cuales son barridos a una trampa primaria donde se acumulan en un absorbente sólido. Cuando se completa la extracción, los COV se retiran de la trampa primaria calentando rápidamente el tubo mientras se enjuaga con He. Esta técnica se conoce como purge-and-trap. Debido a que la recuperación del analito puede no ser reproducible, se requiere un estándar interno para el trabajo cuantitativo.
También se pueden realizar extracciones continuas utilizando fluidos supercríticos [McNally, M. E. Anal. Chem. 1995, 67, 308A-315A]. Si calentamos una sustancia por encima de su temperatura y presión críticas forma un fluido supercrítico cuyas propiedades se encuentran entre las de un gas y un líquido. Un fluido supercrítico es un mejor disolvente que un gas, lo que lo convierte en un mejor reactivo para extracciones. Además, la viscosidad de un fluido supercrítico es significativamente menor que la de un líquido, lo que hace que sea más fácil empujarlo a través de una muestra de partículas. Un ejemplo de extracción de fluido supercrítico es la determinación de hidrocarburos totales de petróleo (TPH) en suelos, sedimentos y lodos usando CO 2 supercrítico [“TPH Extraction by SFE”, ISCO, Inc. Lincoln, NE, Revisado en noviembre de 1992]. Se coloca una muestra de aproximadamente 3 g en un cartucho de acero inoxidable de 10 ml y se pasa CO 2 supercrítico a una presión de 340 atm y una temperatura de 80 o C a través del cartucho durante 30 minutos a un caudal de 1—2 ml/min. Para recoger los TPH, el efluente del cartucho se pasa a través de 3 mL de tetracloroetileno a temperatura ambiente. A esta temperatura el CO 2 vuelve a la fase gaseosa y se libera a la atmósfera.
Separaciones Cromatográficas
En una extracción, la muestra originalmente se encuentra en una fase y extraemos el analito o el interferente en una segunda fase. También podemos separar el analito y los interferentes pasando continuamente una fase libre de muestra, llamada fase móvil, sobre una segunda fase libre de muestra que permanece fija o estacionaria. La muestra se inyecta en la fase móvil y los componentes de la muestra se reparten entre la fase móvil y la fase estacionaria. Aquellos componentes con coeficientes de partición más grandes tienen más probabilidades de pasar a la fase estacionaria y tardan más tiempo en pasar por el sistema. Esta es la base de todas las separaciones cromatográficas. La cromatografía proporciona tanto una separación de analitos e interferentes, como un medio para realizar un análisis cualitativo o cuantitativo para el analito. Por esta razón, en el Capítulo 12 se encuentra un tratamiento más completo de la cromatografía.