
11.4: Métodos voltamétricos y amperométricos
- Page ID
- 75832

En voltamperometría aplicamos un potencial dependiente del tiempo a una celda electroquímica y medimos la corriente resultante en función de ese potencial. Llamamos voltamograma a la gráfica resultante de corriente versus potencial aplicado, y es el equivalente electroquímico de un espectro en espectroscopia, proporcionando información cuantitativa y cualitativa sobre las especies involucradas en la reacción de oxidación o reducción [Maloy, J. T. J. Chem. Educ. 1983, 60, 285—289]. La técnica voltamétrica más temprana es la polarografía, desarrollada por Jaroslav Heyrovsky a principios de la década de 1920, logro por el que fue galardonado con el Premio Nobel de Química en 1959. Desde entonces, se han desarrollado muchas formas diferentes de voltamperometría, algunas de las cuales se destacan en la Figura 11.1.6. Antes de examinar estas técnicas y sus aplicaciones con más detalle, primero debemos considerar el diseño experimental básico para voltamperometría y los factores que influyen en la forma del voltamograma resultante.
Para una introducción en línea a gran parte del material de esta sección, consulte Electroquímica analítica: Los conceptos básicos de Richard S. Kelly, un recurso que forma parte de la Biblioteca Digital de Ciencias Analíticas.
Mediciones Voltamétricas
Aunque los métodos voltamétricos tempranos utilizaron solo dos electrodos, un voltámetro moderno hace uso de un potenciostato de tres electrodos, como el que se muestra en la Figura 11.1.5. En voltamperometría aplicamos una señal de excitación de potencial dependiente del tiempo al electrodo de trabajo, cambiando su potencial en relación con el potencial fijo del electrodo de referencia, y medimos la corriente que fluye entre el electrodo de trabajo y el electrodo auxiliar. El electrodo auxiliar generalmente es un alambre de platino y el electrodo de referencia generalmente es un electrodo SCE o un electrodo de Ag/AgCl.
La Figura 11.1.5 muestra un ejemplo de un potenciostato manual de tres electrodos. Aunque un potenciostato moderno utiliza circuitos muy diferentes, puede usar la Figura 11.1.5 y la discusión que la acompaña para comprender cómo podemos controlar el potencial del electrodo de trabajo y medir la corriente resultante.
Para el electrodo de trabajo podemos elegir entre varios materiales diferentes, incluyendo mercurio, platino, oro, plata y carbono. Las primeras técnicas voltamétricas utilizaron un electrodo de trabajo de mercurio. Debido a que el mercurio es un líquido, el electrodo de trabajo habitual es una gota suspendida del extremo de un tubo capilar. En el electrodo de gota de mercurio colgante, o HMDE, extruimos la gota de Hg girando un tornillo micrómetro que empuja el mercurio desde un depósito a través de un tubo capilar estrecho (Figura 11.4.1 a).
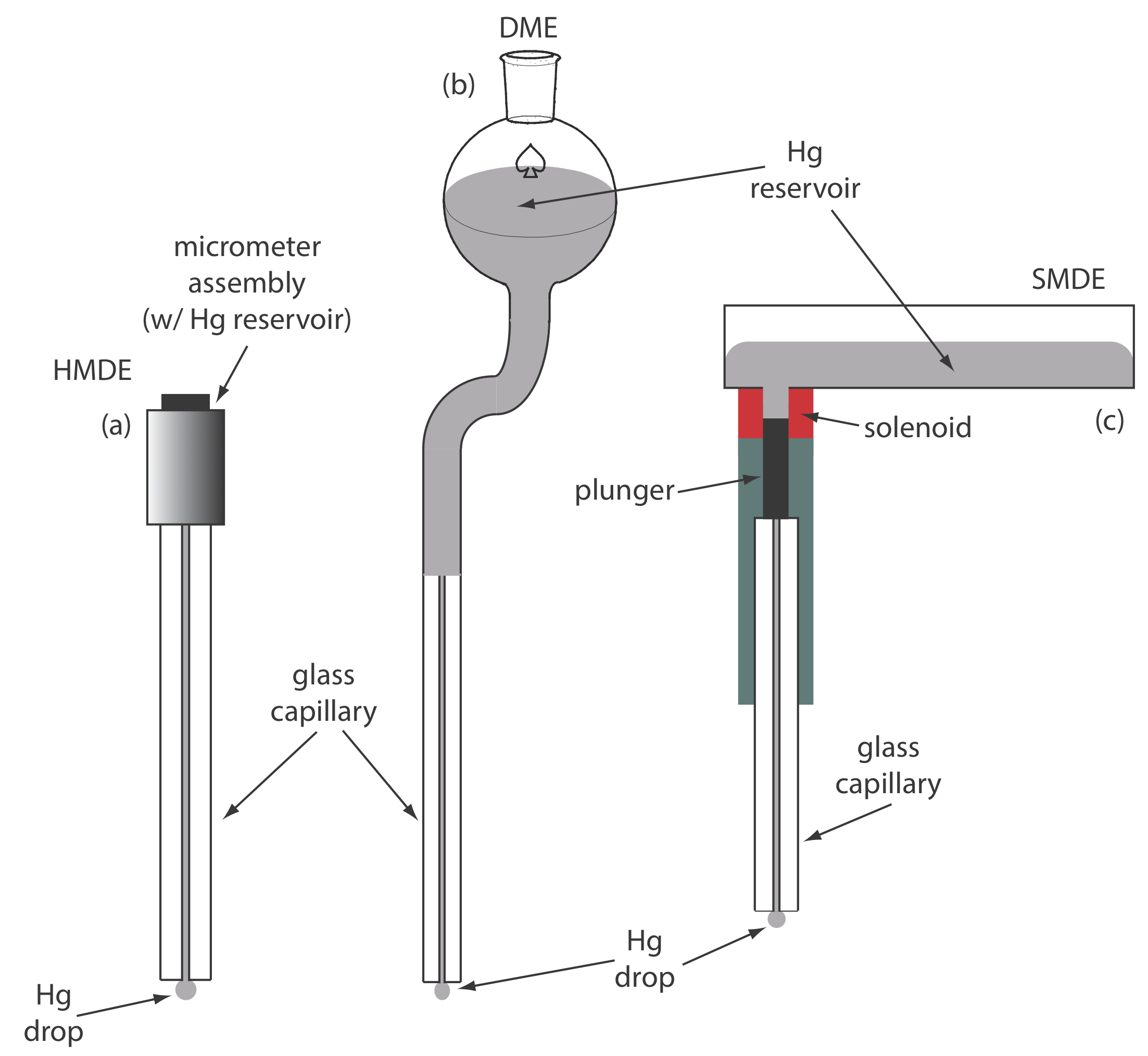
En el electrodo de mercurio que cae, o DME, se forman gotas de mercurio al final del tubo capilar como resultado de la gravedad (Figura 11.4.1 b). A diferencia del HMDE, la gota de mercurio de un DME crece continuamente, a medida que el mercurio fluye del reservorio bajo la influencia de la gravedad, y tiene una vida útil finita de varios segundos. Al final de su vida útil, la gota de mercurio se desaloja, ya sea manualmente o sola, y es reemplazada por una nueva gota. El electrodo estático de gota de mercurio, o SMDE, utiliza un émbolo accionado por solenoide para controlar el flujo de mercurio (Figura 11.4.1 c). La activación del solenoide levanta momentáneamente el émbolo, permitiendo que el mercurio fluya a través del capilar, formando una sola gota de Hg colgante. La activación repetida del solenoide produce una serie de gotas de Hg. De esta manera el SMDE puede ser utilizado ya sea como HMDE o como DME. Hay un tipo adicional de electrodo de mercurio: el electrodo de película de mercurio. Un electrodo sólido, típicamente carbono, platino o oro, se coloca en una solución de Hg 2 + y se mantiene a un potencial donde la reducción de Hg 2 + a Hg es favorable, depositando una película delgada de mercurio sobre la superficie del electrodo sólido.
El mercurio tiene varias ventajas como electrodo de trabajo. Quizás su ventaja más importante es su alto sobrepotencial para la reducción de H 3 O + a H 2, lo que hace que los potenciales accesibles sean tan negativos como —1 V frente al SCE en soluciones ácidas y —2 V frente al SCE en soluciones básicas (Figura 11.4.2 ). Una especie como Zn 2 +, que es difícil de reducir en otros electrodos sin reducir simultáneamente H 3 O +, es fácil de reducir en un electrodo de trabajo de mercurio. Otras ventajas incluyen la capacidad de los metales para disolverse en mercurio, lo que resulta en la formación de una amalgama, y la capacidad de renovar la superficie del electrodo extruyendo una nueva gota. Una limitación al mercurio como electrodo de trabajo es la facilidad con la que se oxida. Dependiendo del solvente, no se puede usar un electrodo de mercurio a potenciales más positivos que aproximadamente —0.3 V a +0.4 V versus el SCE.
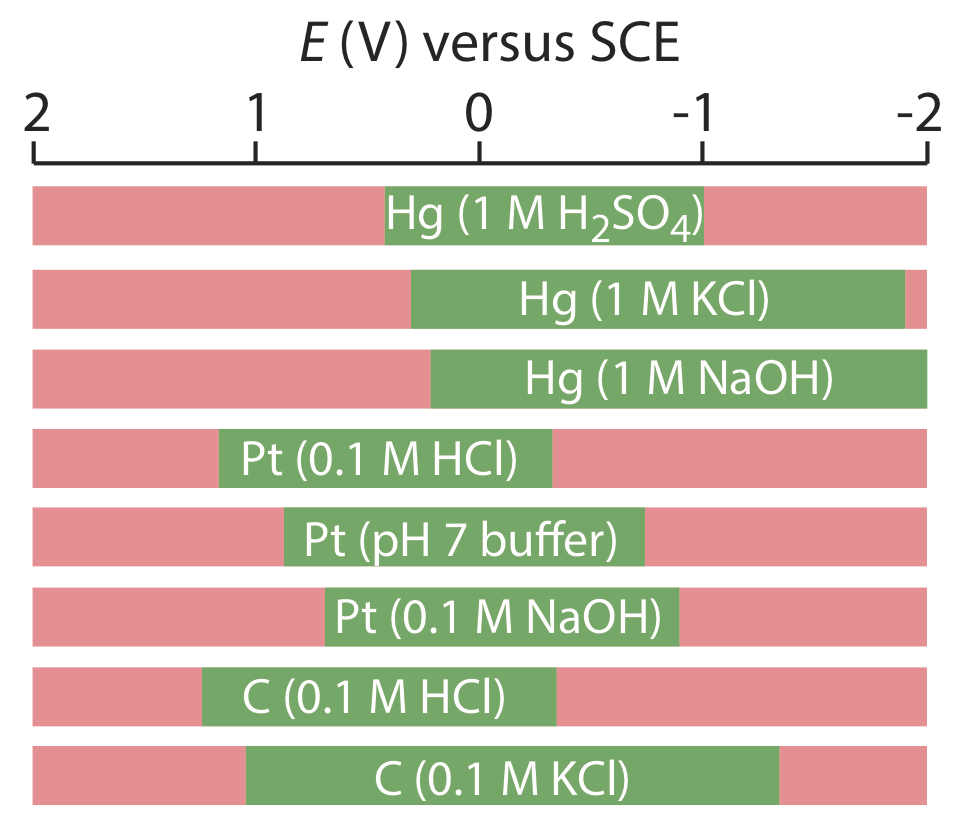
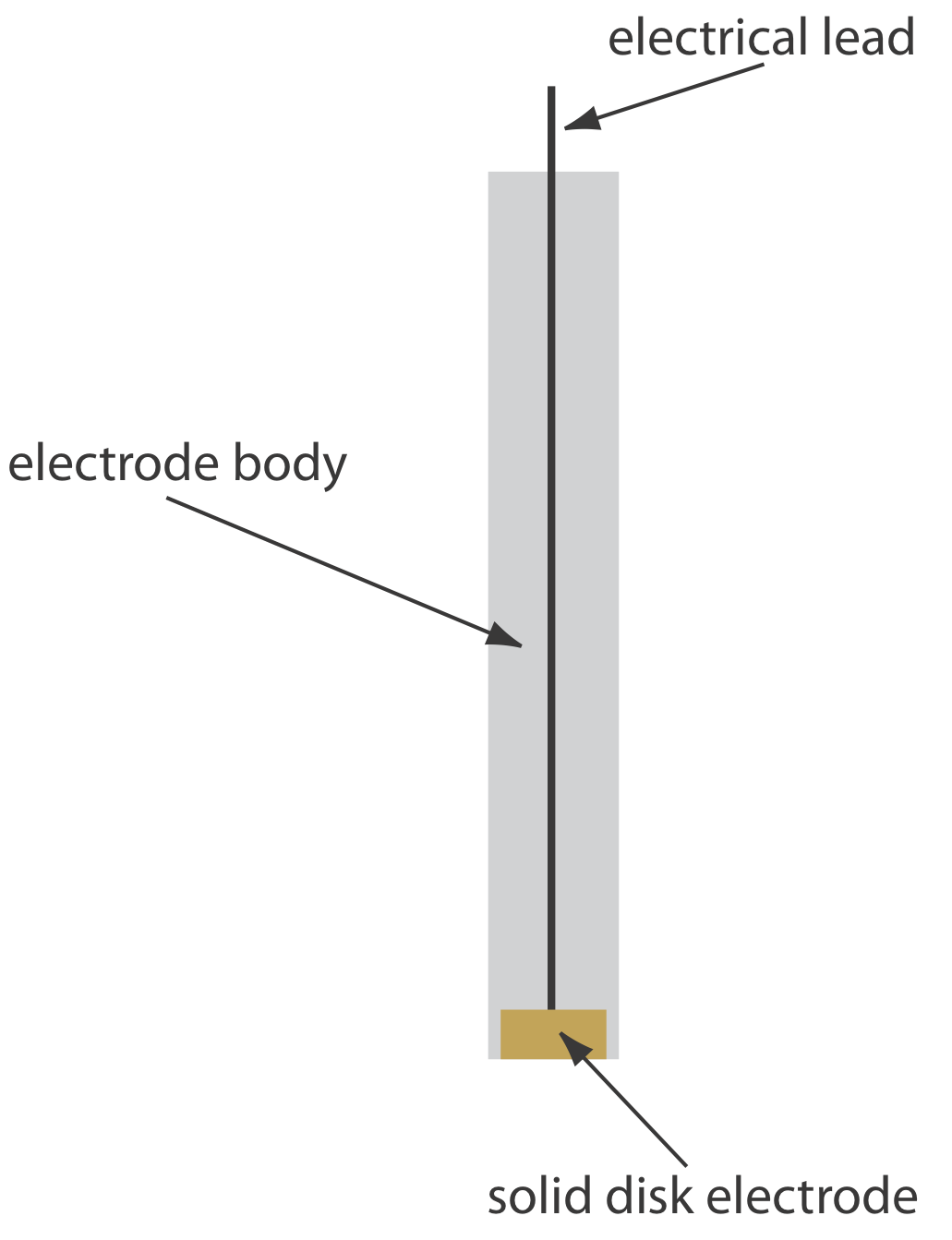
Los electrodos sólidos construidos con platino, oro, plata o carbono se pueden usar en un rango de potenciales, incluyendo potenciales negativos y positivos con respecto al SCE (Figura 11.4.2 ). Por ejemplo, la ventana de potencial para un electrodo de Pt se extiende de aproximadamente +1.2 V a —0.2 V versus el SCE en soluciones ácidas, y de +0.7 V a —1 V versus el SCE en soluciones básicas. Un electrodo sólido puede reemplazar un electrodo de mercurio para muchos análisis voltamétricos que requieren potenciales negativos, y es el electrodo de elección a potenciales más positivos. Excepto por el electrodo de pasta de carbono, un electrodo sólido se forma en un disco y se sella en el extremo de un soporte inerte con un cable eléctrico (Figura 11.4.3 ). El electrodo de pasta de carbono se realiza llenando la cavidad al final del soporte inerte con una pasta que consiste en partículas de carbono y un aceite viscoso. Los electrodos sólidos no están exentos de problemas, el más importante de los cuales es la facilidad con que la superficie del electrodo se ve alterada por la adsorción de una especie de solución o por la formación de una capa de óxido. Por esta razón un electrodo sólido necesita reacondicionamiento frecuente, ya sea mediante la aplicación de un potencial apropiado o por pulido.
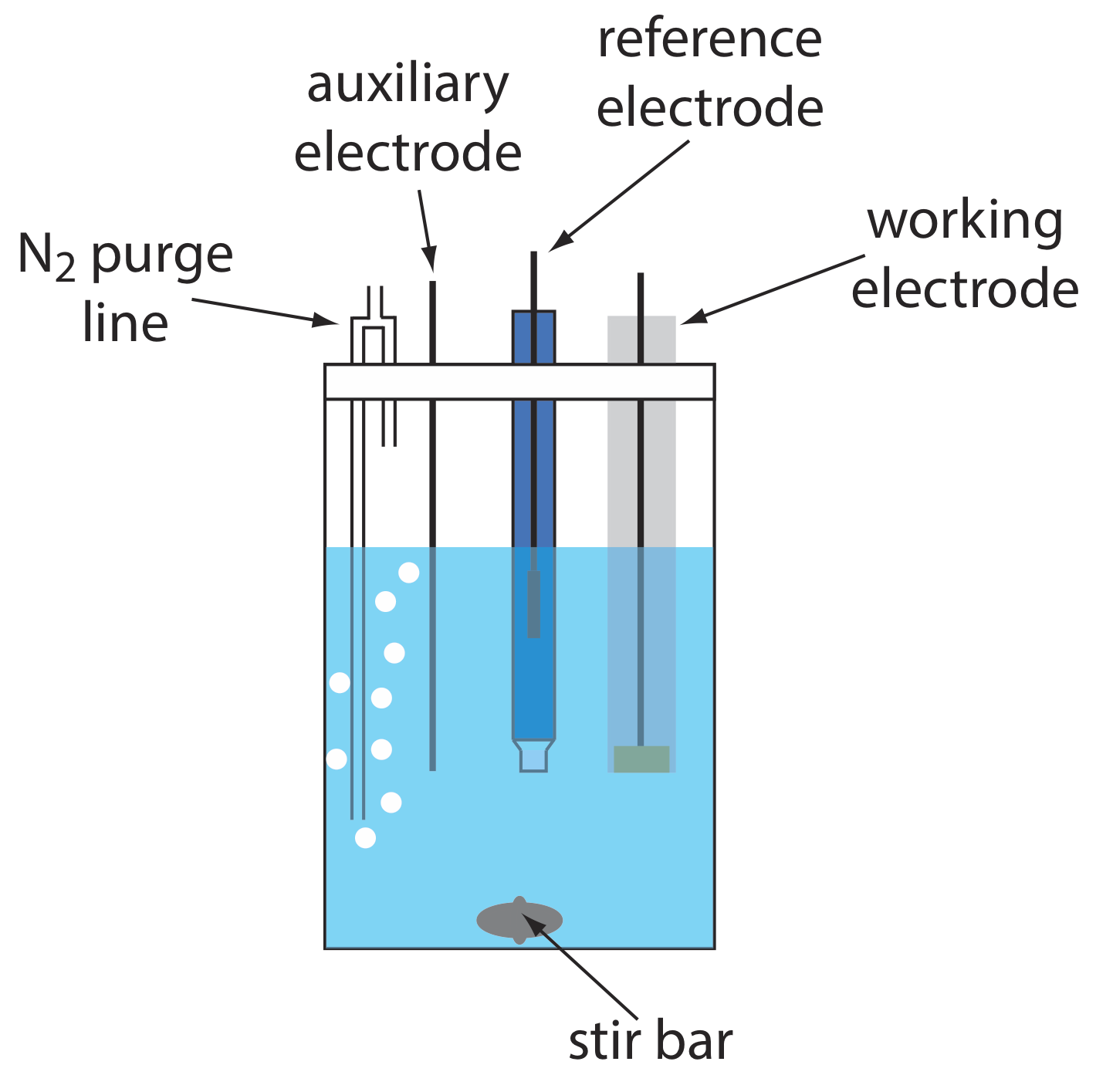
Una disposición típica para una celda electroquímica voltamétrica se muestra en la Figura 11.4.4 . Además del electrodo de trabajo, el electrodo de referencia y el electrodo auxiliar, la celda también incluye una línea de purga N2 para eliminar O 2 disuelto, y una barra de agitación opcional. Las celdas electroquímicas están disponibles en una variedad de tamaños, lo que permite el análisis de volúmenes de solución que van desde más de 100 mL hasta tan pequeños como 50 μL.
Corriente En Voltamperometría
Cuando oxidamos un analito en el electrodo de trabajo, los electrones resultantes pasan a través del potenciostato al electrodo auxiliar, reduciendo el disolvente o algún otro componente de la matriz de solución. Si reducimos el analito en el electrodo de trabajo, la corriente fluye desde el electrodo auxiliar hasta el cátodo. En cualquier caso, la corriente de las reacciones redox en el electrodo de trabajo y los electrodos auxiliares se denomina corriente faradaica. En esta sección consideramos los factores que afectan la magnitud de la corriente faradaica, así como las fuentes de cualquier corriente no faradaica.
Convenciones de Firmar
Debido a que la reacción de interés ocurre en el electrodo de trabajo, describimos la corriente faradaica usando esta reacción. Una corriente faradaica debida a la reducción del analito es una corriente catódica, y su signo es positivo. Una corriente anódica resulta de la oxidación del analito en el electrodo de trabajo, y su signo es negativo.
Influencia del Potencial Aplicado en la Corriente Faradaica
Como ejemplo, consideremos la corriente faradaica cuando reducimos\(\text{Fe(CN)}_6^{3-}\) a\(\text{Fe(CN)}_6^{4-}\) en el electrodo de trabajo. La relación entre las concentraciones de\(\text{Fe(CN)}_6^{3-}\), la concentración y el potencial viene dada por la ecuación de Nernst\(\text{Fe(CN)}_6^{4-}\)
\[E=+0.356 \text{ V}-0.05916 \log \frac{\left[\mathrm{Fe}(\mathrm{CN})_{6}^{4-}\right]_{x=0}}{\left[\mathrm{Fe}(\mathrm{CN})_{6}^{3-}\right]_{x=0}} \nonumber\]
donde +0.356V es el estadoestándar-potencial para el par\(\text{Fe(CN)}_6^{3-}\)/\(\text{Fe(CN)}_6^{4-}\)redox, y x = 0 indica que las concentraciones de\(\text{Fe(CN)}_6^{3-}\) - y\(\text{Fe(CN)}_6^{4-}\) son aquellas en la superficie del electrodo de trabajo. Utilizamos concentraciones superficiales en lugar de concentraciones a granel porque la posición de equilibrio para la reacción redox
\[\mathrm{Fe}(\mathrm{CN})_{6}^{3-}(a q)+e^{-}\rightleftharpoons\mathrm{Fe}(\mathrm{CN})_{6}^{4-}(a q) \nonumber\]
se establece en la superficie del electrodo.
Supongamos que tenemos una solución para la cual la concentración inicial de\(\text{Fe(CN)}_6^{3-}\) es 1.0 mM y que\(\text{Fe(CN)}_6^{4-}\) está ausente. La figura 11.4.5 muestra el diagrama de escalera para esta solución.
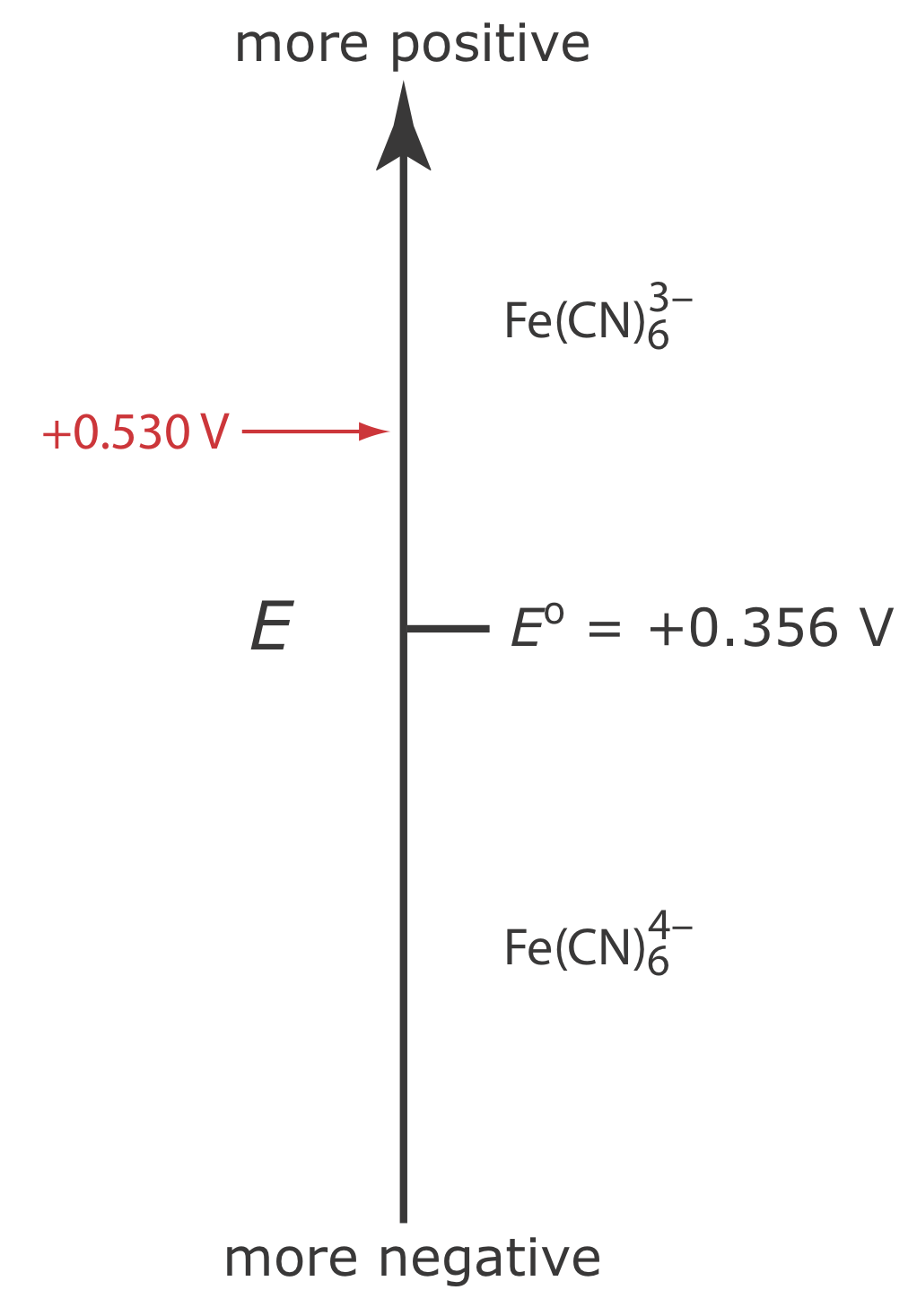
Si aplicamos un potencial de +0.530 V al electrodo de trabajo, las concentraciones de\(\text{Fe(CN)}_6^{3-}\) y\(\text{Fe(CN)}_6^{4-}\) en la superficie del electrodo no se ven afectadas, y no se observa corriente faradaica. Si cambiamos el potencial a +0.356 V parte del\(\text{Fe(CN)}_6^{3-}\) en la superficie del electrodo se reduce\(\text{Fe(CN)}_6^{4-}\) hasta llegar a una condición en la que
\[\left[\mathrm{Fe}(\mathrm{CN})_{6}^{3-}\right]_{x=0}=\left[\mathrm{Fe}(\mathrm{CN})_{6}^{4-}\right]_{x=0}=0.50 \text{ mM} \nonumber\]
Este es el primero de los cinco principios importantes de la electroquímica descritos en el Capítulo 11.1: el potencial del electrodo determina la forma del analito en la superficie del electrodo.
Si esto es todo lo que sucede después de aplicar el potencial, entonces habría una breve oleada de corriente faradaica que rápidamente vuelve a cero, que no es el más interesante de los resultados. Aunque las concentraciones de\(\text{Fe(CN)}_6^{3-}\) y\(\text{Fe(CN)}_6^{4-}\) en la superficie del electrodo son 0.50 mM, sus concentraciones en solución a granel permanecen sin cambios.
Este es el segundo de los cinco principios importantes de la electroquímica esbozados en el Capítulo 11.1: la concentración del analito en el electrodo puede no ser la misma que su concentración en solución a granel.
Debido a esta diferencia de concentración, existe un gradiente de concentración entre la solución en la superficie del electrodo y la solución a granel. Este gradiente de concentración crea una fuerza impulsora que\(\text{Fe(CN)}_6^{4-}\) se aleja del electrodo y que se transporta\(\text{Fe(CN)}_6^{3-}\) al electrodo (Figura 11.4.6 ). A medida que el\(\text{Fe(CN)}_6^{3-}\) llega al electrodo, también, se reduce a\(\text{Fe(CN)}_6^{4-}\). Una corriente faradaica continúa fluyendo hasta que no hay diferencia entre las concentraciones de\(\text{Fe(CN)}_6^{3-}\) y\(\text{Fe(CN)}_6^{4-}\) en el electrodo y sus concentraciones en solución a granel.
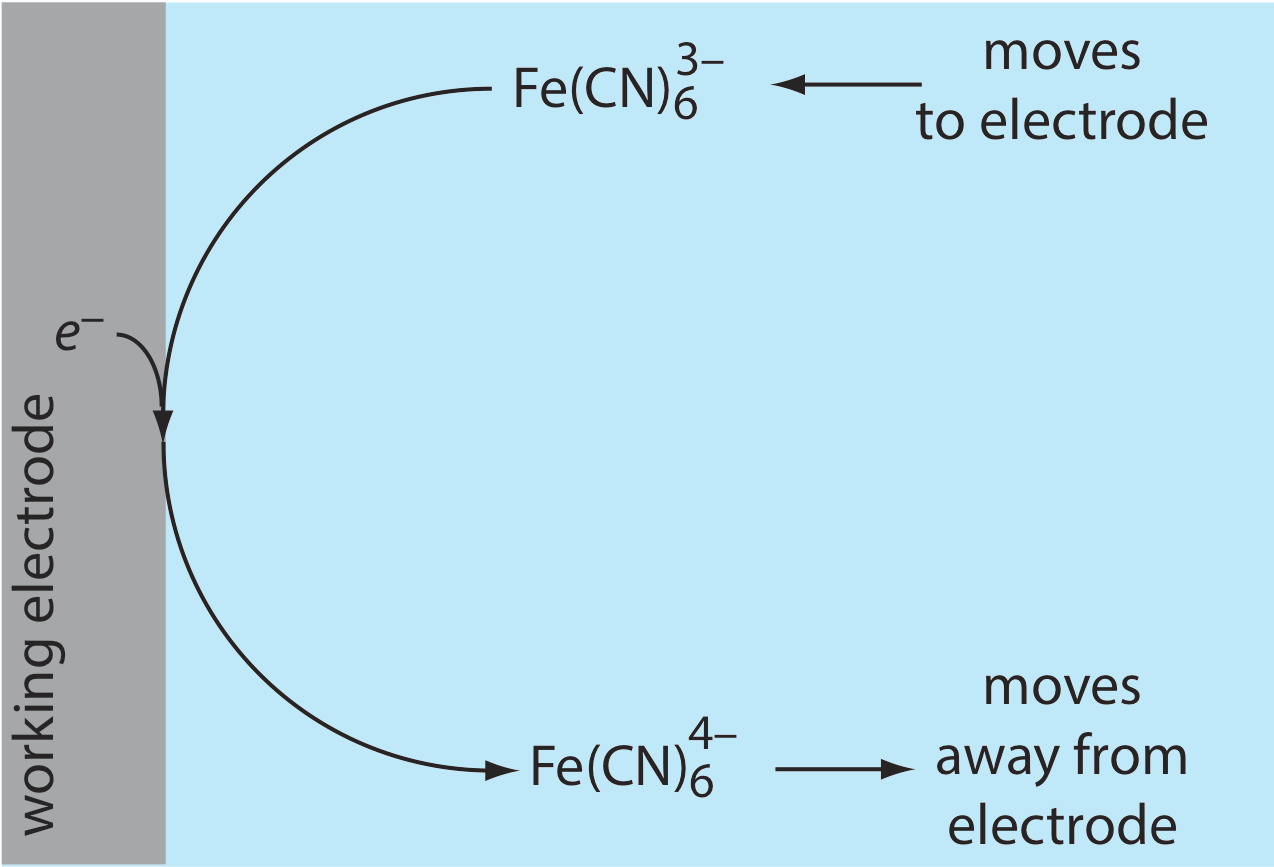
Aunque el potencial en el electrodo de trabajo determina si fluye una corriente faradaica, la magnitud de la corriente está determinada por la velocidad de la reacción de oxidación o reducción resultante. Dos factores contribuyen a la velocidad de la reacción electroquímica: la velocidad a la que los reactivos y productos son transportados hacia y desde el electrodo, lo que llamamos transporte de masa, y la velocidad a la que los electrones pasan entre el electrodo y los reactivos y productos en solución.
Este es el cuarto de los cinco principios importantes de la electroquímica esbozados en el Capítulo 11.1: la corriente es una medida de velocidad.
Influencia del transporte masivo en la corriente faradaica
Hay tres modos de transporte de masa que afectan la velocidad a la que los reactivos y productos se mueven hacia o lejos de la superficie del electrodo: difusión, migración y convección. La difusión ocurre siempre que la concentración de un ion o una molécula en la superficie del electrodo es diferente de la de la solución a granel. Si aplicamos un potencial suficiente para reducir completamente\(\text{Fe(CN)}_6^{3-}\) en la superficie del electrodo, el resultado es un gradiente de concentración similar al mostrado en la Figura 11.4.7 . La región de solución sobre la que se produce la difusión es la capa de difusión. A falta de otros modos de transporte masivo, el ancho de la capa de difusión,\(\delta\), aumenta con el tiempo ya que el\(\text{Fe(CN)}_6^{3-}\) mosto se difunde desde una distancia cada vez mayor.
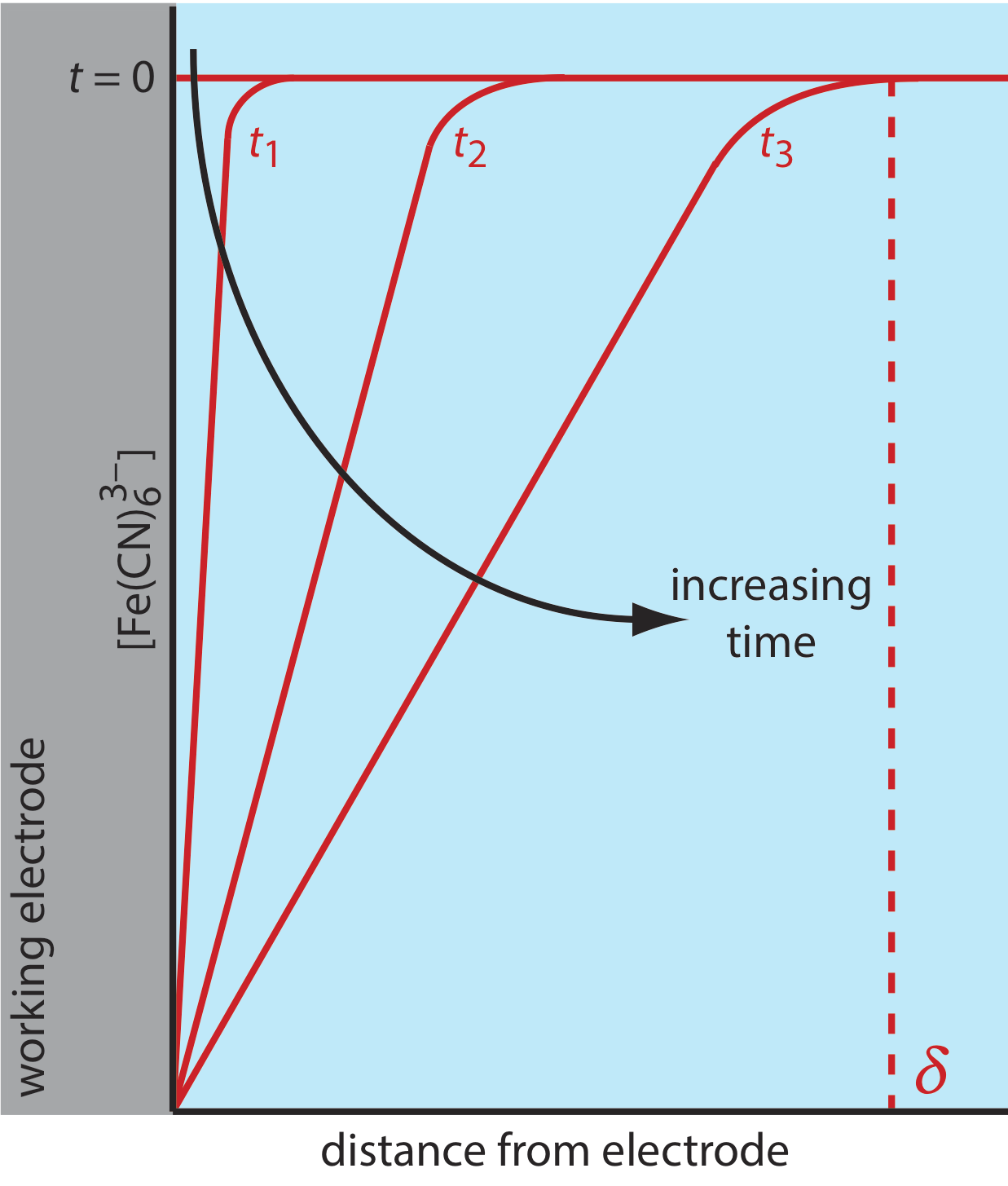
La convección ocurre cuando mezclamos la solución, que lleva los reactivos hacia el electrodo y elimina los productos del electrodo. La forma más común de convección es agitar la solución con una barra agitadora; otros métodos incluyen rotar el electrodo e incorporar el electrodo a una celda de flujo.
El modo final de transporte de masa es la migración, que ocurre cuando una partícula cargada en solución es atraída o repelida de un electrodo que porta una carga superficial. Si el electrodo lleva una carga positiva, por ejemplo, un anión se moverá hacia el electrodo y un catión se moverá hacia la solución a granel. A diferencia de la difusión y convección, la migración afecta únicamente al transporte masivo de partículas cargadas.
El movimiento del material hacia y desde la superficie del electrodo es una función compleja de los tres modos de transporte de masa. En el límite donde la difusión es la única forma significativa de transporte de masa, la corriente en una celda voltamétrica es igual a
\[i=\frac{n F A D\left(C_{\text {bulk }}-C_{x=0}\right)}{\delta} \label{11.1}\]
donde n el número de electrones en la reacción redox, F es la constante de Faraday, A es el área del electrodo, D es el coeficiente de difusión para las especies que reaccionan en el electrodo, C a granel y C x = 0 son sus concentraciones en solución a granel y en la superficie del electrodo, y\(\delta\) es el grosor de la capa de difusión.
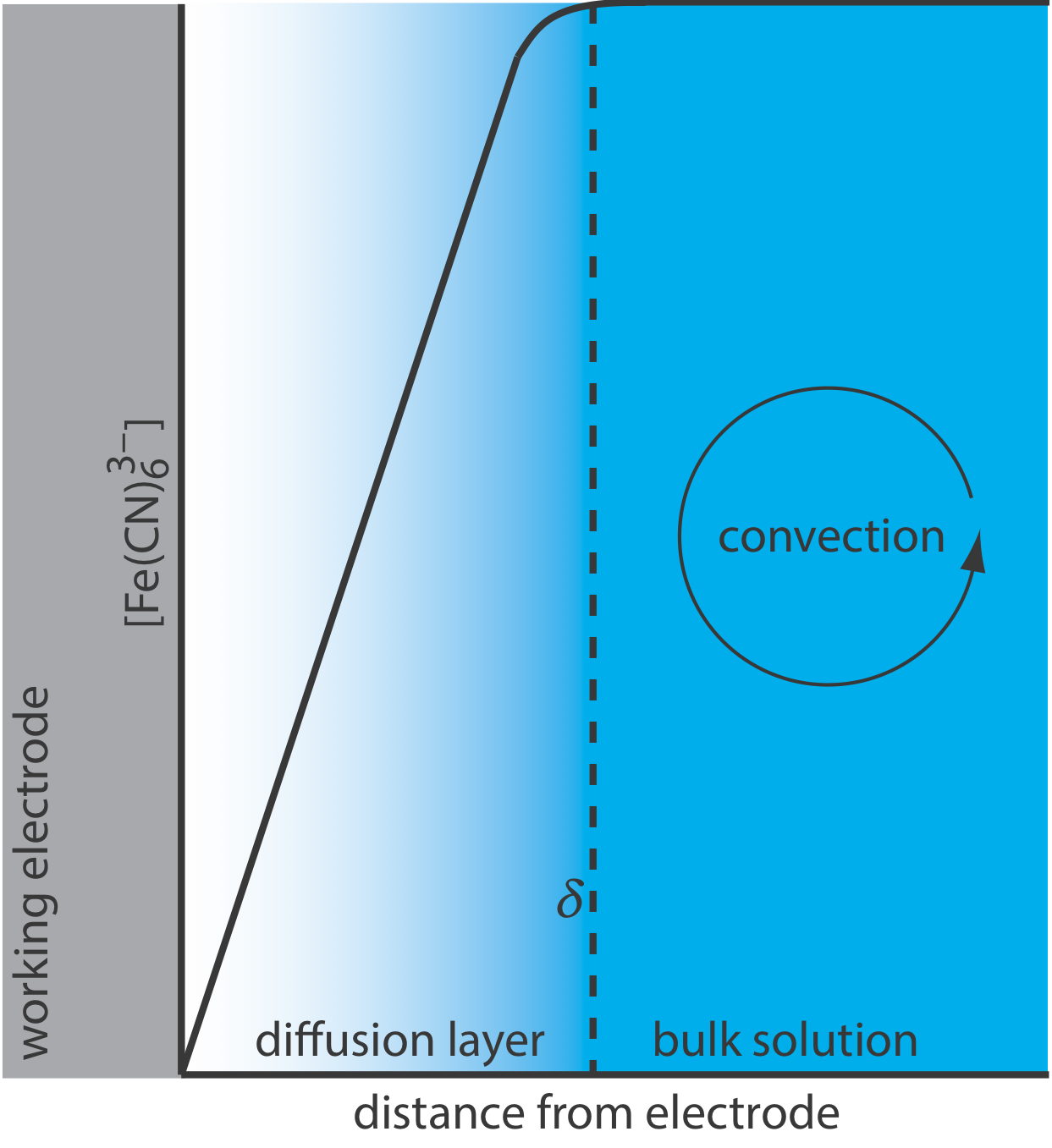
Para que la Ecuación\ ref {11.1} sea válida, la convección y migración no deben interferir con la formación de una capa de difusión. Podemos eliminar la migración agregando una alta concentración de un electrolito de soporte inerte. Debido a que los iones de carga similar son igualmente atraídos o repelidos desde la superficie del electrodo, cada uno tiene la misma probabilidad de sufrir migración. Un gran exceso de electrolito inerte asegura que pocos reactivos o productos experimenten migración. Si bien es fácil eliminar la convección al no agitar la solución, existen diseños experimentales donde no podemos evitar la convección, ya sea porque debemos agitar la solución o porque estamos usando una celda de flujo electroquímica. Afortunadamente, como se muestra en la Figura 11.4.8 , la dinámica de un fluido que se mueve más allá de un electrodo da como resultado una pequeña capa de difusión, típicamente de 1—10 μm de grosor, en la que la tasa de transporte de masa por convección cae a cero.
Efecto de la cinética de transferencia de electrones sobre la corriente faradaica
La tasa de transporte de masa es un factor que influye en la corriente en voltamperometría. La facilidad con la que los electrones se mueven entre el electrodo y las especies que reaccionan en el electrodo también afecta a la corriente. Cuando la cinética de transferencia de electrones es rápida, la reacción redox está en equilibrio. En estas condiciones la reacción redox es electroquímicamente reversible y se aplica la ecuación de Nernst. Si la cinética de transferencia de electrones es suficientemente lenta, la concentración de reactivos y productos en la superficie del electrodo y, por lo tanto, la magnitud de la corriente faradaica, no son lo que predice la ecuación de Nernst. En este caso el sistema es electroquímicamente irreversible.
Corrientes de Carga
Además de la corriente faradaica de una reacción redox, la corriente en una celda electroquímica incluye otras fuentes no faradáicas. Supongamos que la carga en un electrodo es cero y de repente cambiamos su potencial para que la superficie del electrodo adquiera una carga positiva. Los cationes cerca de la superficie del electrodo responderán a esta carga positiva migrando lejos del electrodo; los aniones, por otro lado, migrarán hacia el electrodo. Esta migración de iones se produce hasta que la carga superficial positiva del electrodo y la carga negativa de la solución cerca del electrodo son iguales. Debido a que el movimiento de los iones y el movimiento de los electrones son indistinguibles, el resultado es una pequeña corriente no faradaica de corta duración que llamamos corriente de carga. Cada vez que cambiamos el potencial del electrodo, fluye una corriente de carga transitoria.
La migración de iones en respuesta a la carga superficial del electrodo conduce a la formación de una interfaz estructurada electrodo-solución que llamamos la doble capa eléctrica, o EDL. Cuando cambiamos el potencial de un electrodo, la corriente de carga es el resultado de una reestructuración de la EDL. La estructura exacta de la doble capa eléctrica no es importante en el contexto de este texto, pero puede consultar los recursos adicionales de este capítulo para obtener información adicional.
Corriente Residual
Incluso en ausencia de analito, una pequeña corriente medible fluye a través de una celda electroquímica. Esta corriente residual tiene dos componentes: una corriente faradaica debida a la oxidación o reducción de impurezas traza y una corriente de carga no faradaica. Los métodos para discriminar entre la corriente faradaica del analito y la corriente residual se discuten más adelante en este capítulo.
Forma de Voltamogramas
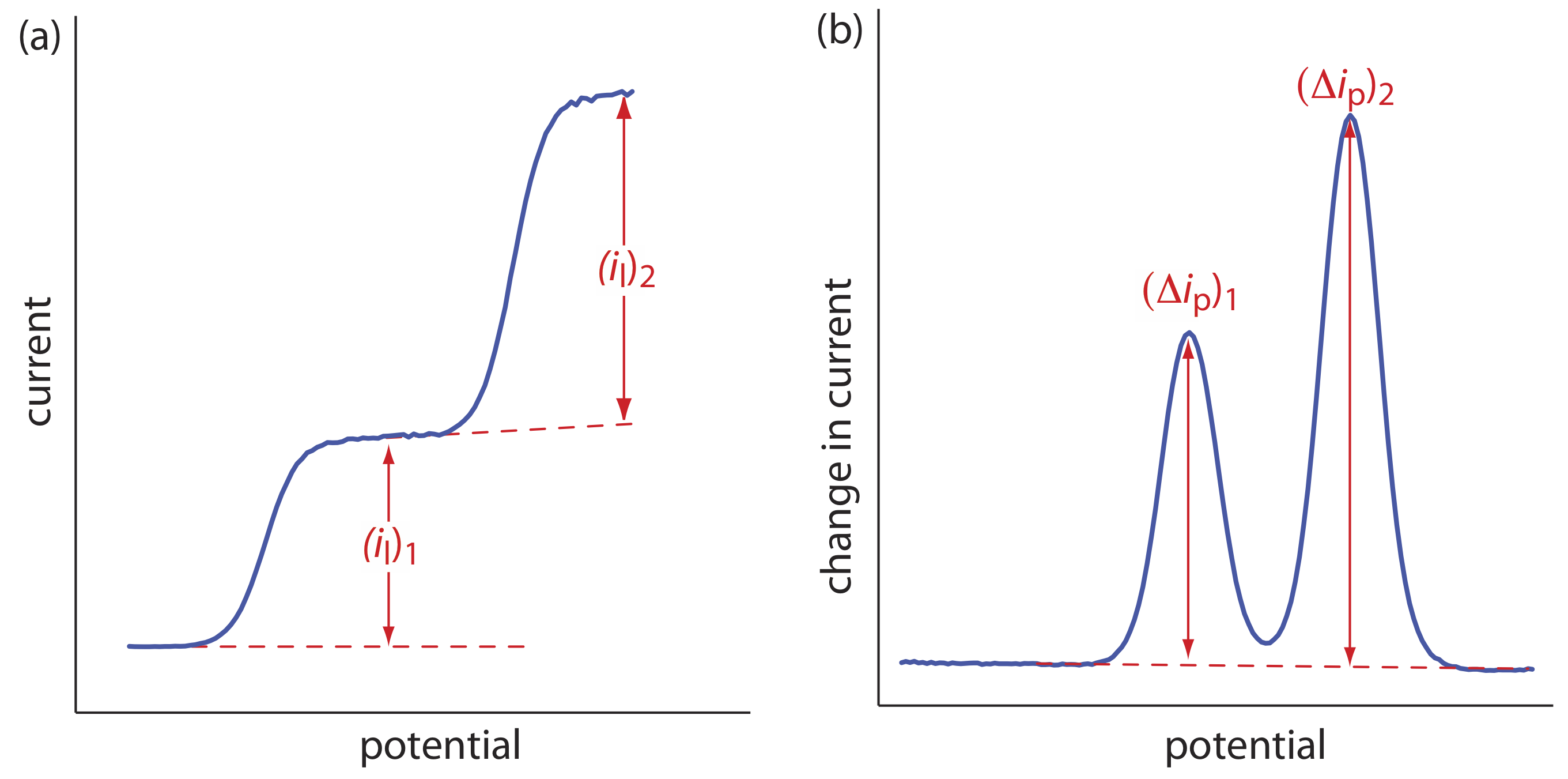
La forma de un voltamograma está determinada por varios factores experimentales, el más importante de los cuales es cómo medimos la corriente y si la convección se incluye como medio de transporte masivo. Como se muestra en la Figura 11.4.9 , a pesar de la abundancia de diferentes técnicas voltamétricas, varias de las cuales se discuten en este capítulo, solo hay tres formas comunes para los voltammogramas.
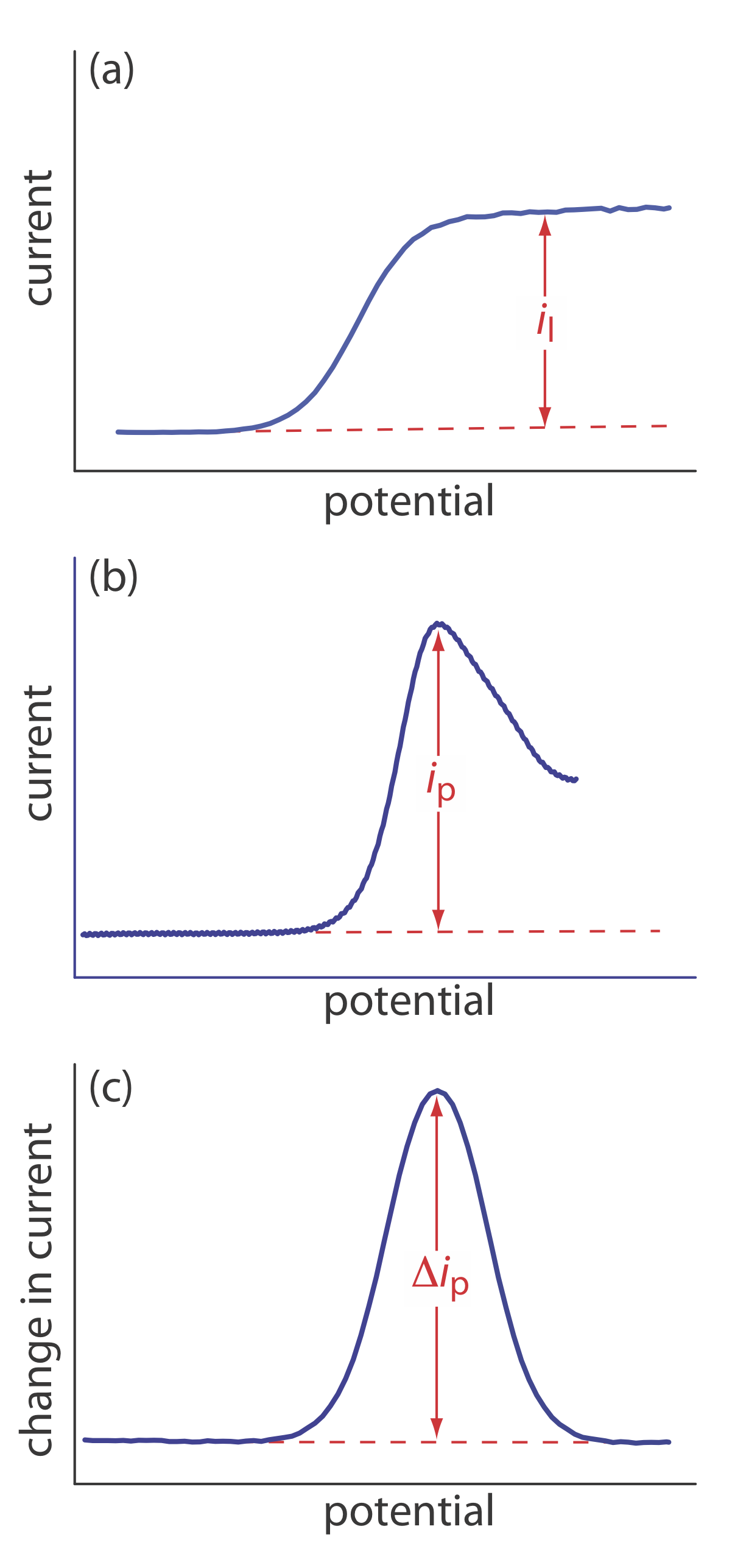
Para el voltammograma de la Figura 11.4.9 a, la corriente aumenta de una corriente residual de fondo a una corriente limitante, i l. Debido a que la corriente faradaica es inversamente proporcional a\(\delta\) (Ecuación\ ref {11.1}), se produce una corriente limitante solo si el espesor de la capa de difusión permanece constante porque estamos agitando la solución (ver Figura 11.4.8 ). En ausencia de convección la capa de difusión aumenta con el tiempo (ver Figura 11.4.7 ). Como se muestra en la Figura 11.4.9 b, el voltamograma resultante tiene una corriente pico en lugar de una corriente limitante.
Para los voltammogramas de la Figura 11.4.9 a y la Figura 11.4.9 b, medimos la corriente en función del potencial aplicado. También podemos monitorear el cambio en la corriente,\(\Delta i\), luego de un cambio en el potencial. El voltamograma resultante, mostrado en la Figura 11.4.9 c, también tiene una corriente pico.
Aspectos cuantitativos y cualitativos de la voltametría
Anteriormente describimos un voltamograma como el equivalente electroquímico de un espectro en espectroscopía. En esta sección consideramos cómo podemos extraer información cuantitativa y cualitativa a partir de un voltamograma. Por simplicidad limitaremos nuestro tratamiento a voltammogramas similares a la Figura 11.4.9 a.
Determinar la concentración
Supongamos que la reacción redox en el electrodo de trabajo es
\[O+n e^{-} \rightleftharpoons R \label{11.2}\]
donde O es la forma oxidada del analito y R es su forma reducida. Supongamos también que solo O inicialmente está presente en solución a granel y que estamos agitando la solución. Cuando aplicamos un potencial que da como resultado la reducción de O a R, la corriente depende de la velocidad a la que O se difunde a través de la capa de difusión fija que se muestra en la Figura 11.4.7 . Usando la ecuación\ ref {11.1}, la corriente, i, es
\[i=K_{O}\left([O]_{\text {bulk }}-[O]_{x=0}\right) \label{11.3}\]
donde K O es una constante igual a\(n F A D_O / \delta\). Cuando alcanzamos la corriente limitante, i l, la concentración de O en la superficie del electrodo es cero y la Ecuación\ ref {11.3} simplifica a
\[i_{l}=K_{O}[O]_{\mathrm{bulk}} \label{11.4}\]
La ecuación\ ref {11.4} nos muestra que la corriente limitante es una función lineal de la concentración de O en solución a granel. Para determinar el valor de K O podemos utilizar cualquiera de los métodos de estandarización cubiertos en el Capítulo 5. Ecuaciones similares a la Ecuación\ ref {11.4} se pueden desarrollar para los otros dos tipos de voltammogramas mostrados en la Figura 11.4.9 .
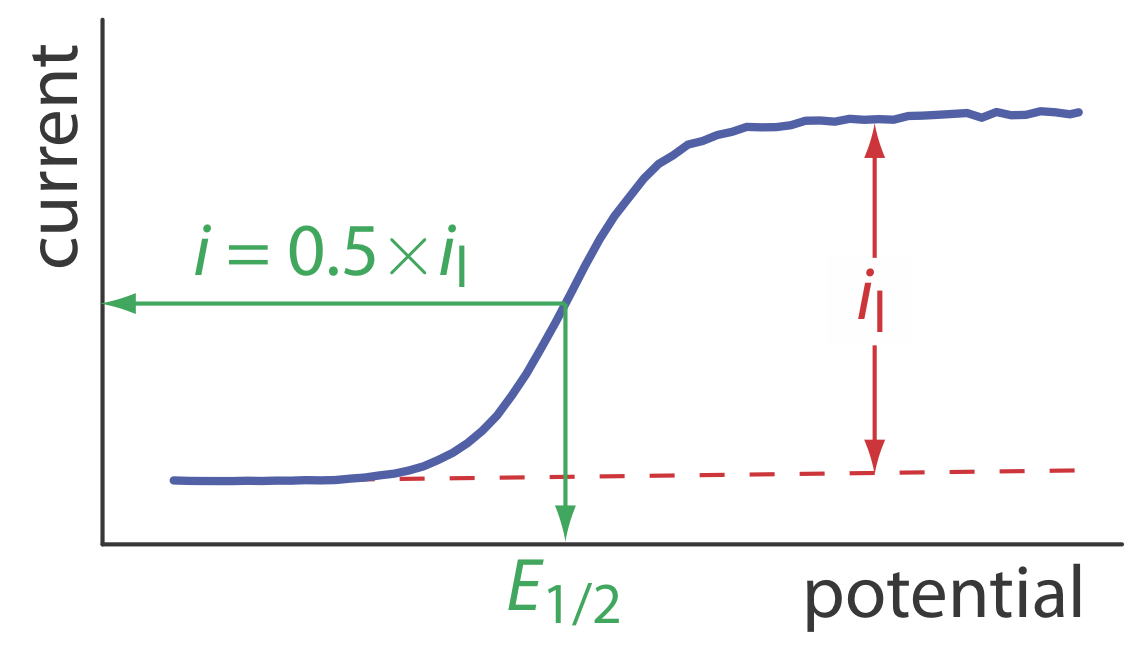
Determinar el potencial de estado estándar
Para extraer el potencial de estado estándar de un voltamograma, necesitamos reescribir la ecuación de Nernst para la reacción\ ref {11.2}
\[E=E_{O / R}^{\circ}-\frac{0.05916}{n} \log \frac{[R]_{x=0}}{[O]_{x=0}} \label{11.5}\]
en términos de corriente en lugar de las concentraciones de O y R. Esto lo haremos en varios pasos. Primero, sustituimos la Ecuación\ ref {11.4} en la Ecuación\ ref {11.3} y reorganizamos para dar
\[[O]_{x=0}=\frac{i_{l}-i}{K_{O}} \label{11.6}\]
A continuación, derivamos una ecuación similar para [R] x = 0, señalando que
\[i=K_{R}\left([R]_{x=0}-[R]_{\mathrm{bulk}}\right) \nonumber\]
Debido a que la concentración de [R] bulk es cero, recuerde nuestra suposición de que la solución inicial contiene solo O, podemos simplificar esta ecuación
\[i=K_{R}[R]_{x=0} \nonumber\]
y resolver para [R] x = 0.
\[[R]_{x=0}=\frac{i}{K_{R}} \label{11.7}\]
Ahora estamos listos para terminar nuestra derivación. Sustituyendo la Ecuación\ ref {11.7} y la Ecuación\ ref {11.6} en la Ecuación\ ref {11.5} y el reordenamiento nos deja con
Cuando la corriente, i, es la mitad de la corriente limitante, i l,
\[i=0.5 \times i_{l} \nonumber\]
podemos simplificar la Ecuación\ ref {11.8} a
\[E_{1 / 2}=E_{O / R}^{\circ}-\frac{0.05916}{n} \log \frac{K_{O}}{K_{R}} \label{11.9}\]
donde E 1/2 es el potencial de media onda (Figura 11.4.10 ). Si K O es aproximadamente igual a K R, que a menudo es el caso, entonces el potencial de media onda es igual al potencial de estado estándar. Tenga en cuenta que la Ecuación\ ref {11.9} es válida solo si la reacción redox es electroquímicamente reversible.
Técnicas Voltamétricas
En voltamperometría hay tres parámetros experimentales importantes bajo nuestro control: cómo cambiamos el potencial aplicado al electrodo de trabajo, cuándo elegimos medir la corriente, y si elegimos agitar la solución. No es sorprendente que existan muchas técnicas voltamétricas diferentes. En esta sección consideramos varios ejemplos importantes.
Polarografía
La primera técnica voltamétrica importante a desarrollar, la polarografía, utiliza como electrodo de trabajo el electrodo de mercurio que se muestra en la Figura 11.4.1 b. Como se muestra en la Figura 11.4.11 , la corriente se mide mientras se aplica una rampa de potencial lineal.
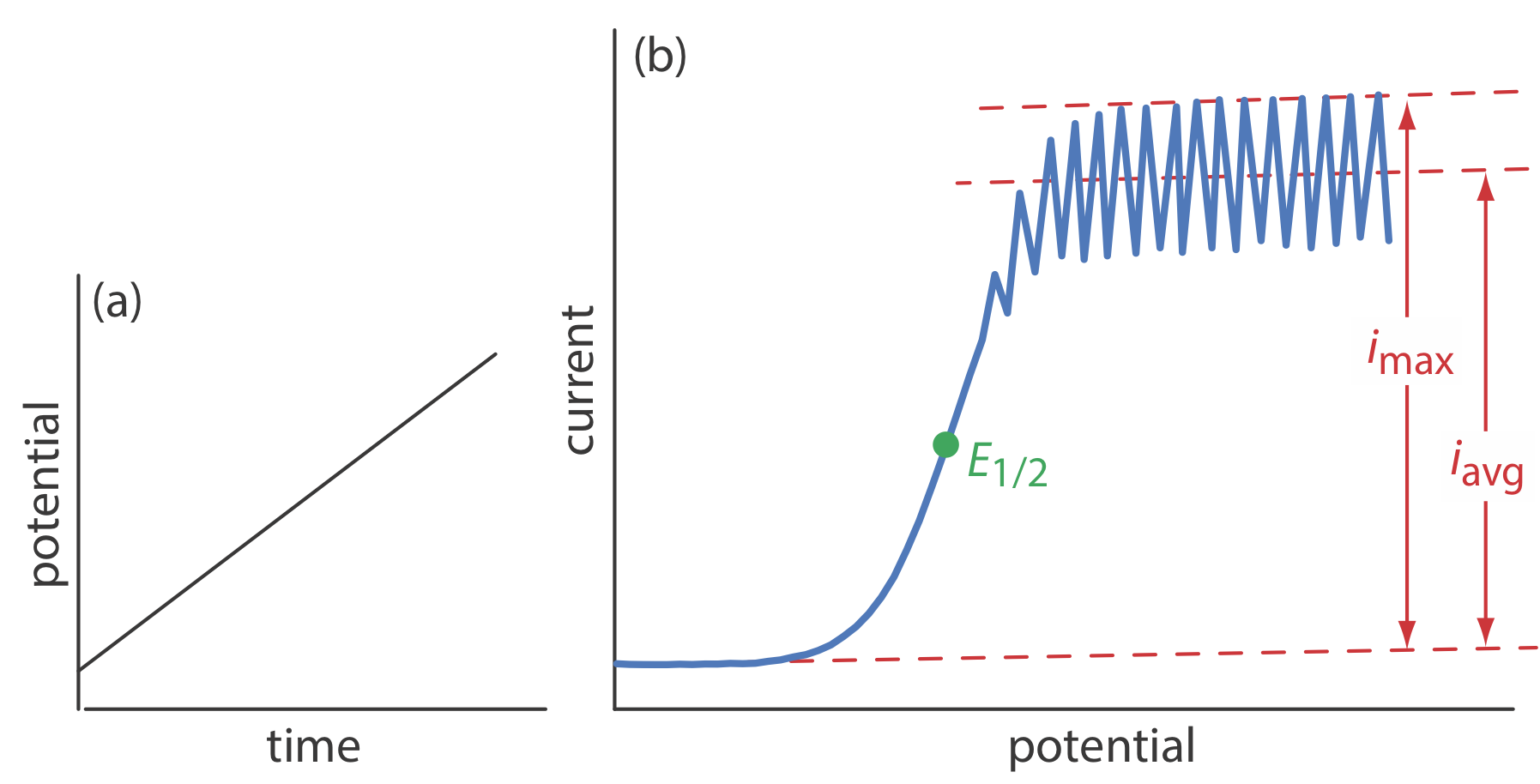
Aunque la polarografía se realiza en una solución sin agitación, obtenemos una corriente limitante en lugar de una corriente pico. Cuando una gota de Hg se separa del capilar de vidrio y cae al fondo de la celda electroquímica, mezcla la solución. Cada nueva gota de Hg, por lo tanto, se convierte en una solución cuya composición es idéntica a la solución a granel. Las oscilaciones en la corriente son el resultado del crecimiento de la caída de Hg, lo que conduce a un cambio dependiente del tiempo en el área del electrodo de trabajo. La corriente limitante, que también se llama corriente de difusión, se mide usando la corriente máxima, i max, o a partir de la corriente promedio, i avg. La relación entre la concentración del analito, C A, y la corriente limitante viene dada por las ecuaciones de Ilkovic
\[i_{\max }=706 n D^{1 / 2} m^{2 / 3} t^{1 / 6} C_{A}=K_{\max } C_{A} \nonumber\]
\[i_{avg}=607 n D^{1 / 2} m^{2 / 3} t^{1 / 6} C_{A}=K_{\mathrm{avg}} C_{A} \nonumber\]
donde n es el número de electrones en la reacción redox, D es el coeficiente de difusión del analito, m es el caudal de Hg, t es la vida útil de la gota y K max y K avg son constantes. El potencial de media onda, E 1/2, proporciona información cualitativa sobre la reacción redox.
La polarografía normal ha sido reemplazada por diversas formas de polarografía de pulso, varios ejemplos de las cuales se muestran en la Figura 11.4.12 [Osteryoung, J. J. Chem. Educ. 1983, 60, 296—298]. Polarografía de pulso normal (Figura 11.4.12 a), por ejemplo, utiliza una serie de pulsos potenciales caracterizados por un ciclo de tiempo\(\tau\), un pulso-tiempo de t p, un potencial de pulso de\(\Delta E_\text{p}\), y un cambio en el potencial por ciclo de\(\Delta E_\text{s}\). Las condiciones experimentales típicas para la polarografía de pulso normal son\(\tau \approx 1 \text{ s}\), t p ≈ 50 ms, y\(\Delta E_\text{s} \approx 2 \text{ mV}\). El valor inicial de\(\Delta E_\text{p} \approx 2 \text{ mV}\), y aumenta en ≈ 2 mV con cada pulso. La corriente se muestrea al final de cada pulso de potencial durante aproximadamente 17 ms antes de devolver el potencial a su valor inicial. La forma del voltamograma resultante es similar a la Figura 11.4.11 , pero sin las oscilaciones actuales. Debido a que aplicamos el potencial para solo una pequeña porción de la vida útil de la gota, hay menos tiempo para que el analito sufra oxidación o reducción y una capa de difusión más pequeña. Como resultado, la corriente faradaica en la polarografía de pulso normal es mayor que en la polarografía, resultando en una mejor sensibilidad y menores límites de detección.
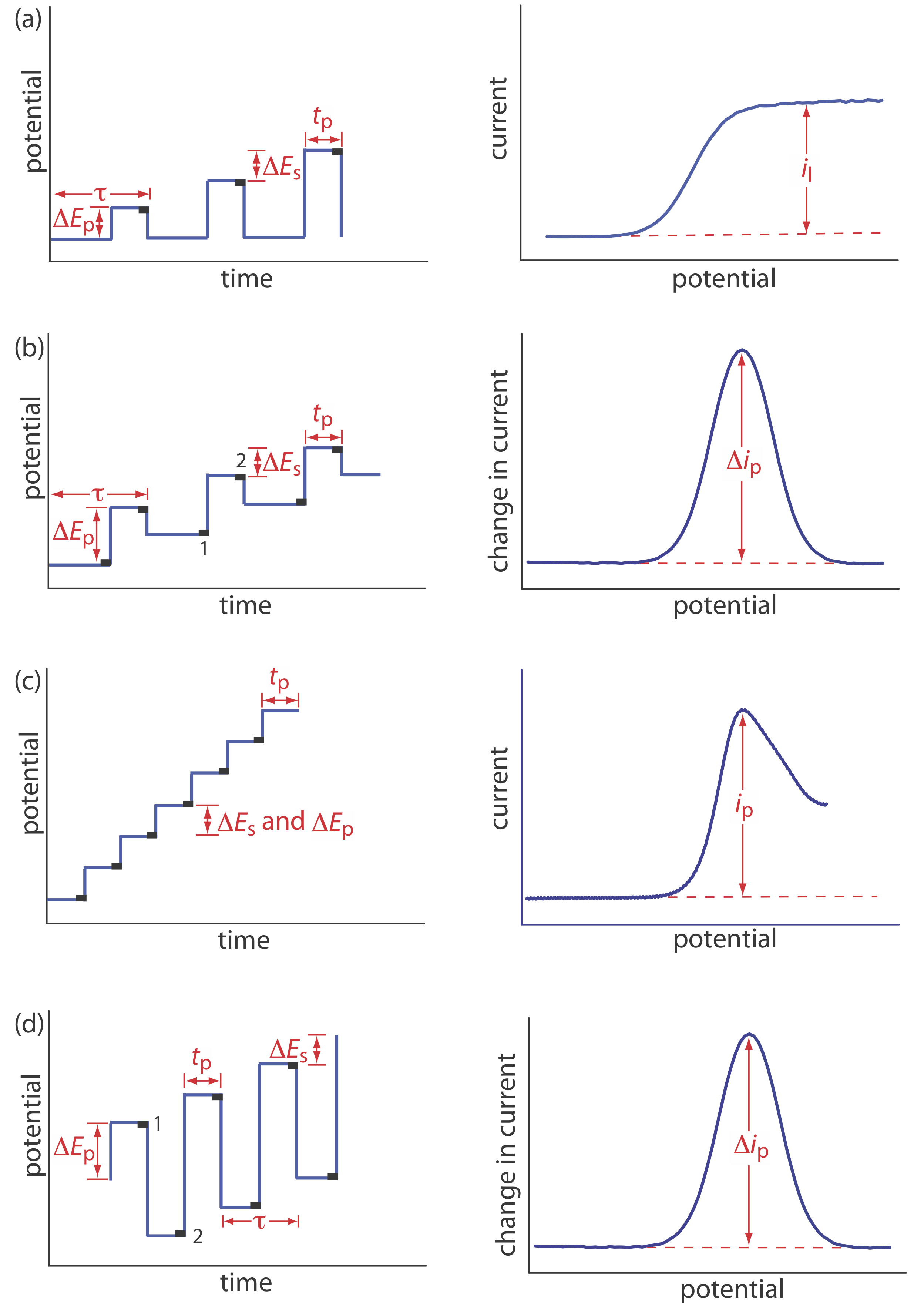
En la polarografía diferencial de pulsos (Figura 11.4.12 b) la corriente se mide dos veces por ciclo: por aproximadamente 17 ms antes de aplicar el pulso y por aproximadamente 17 ms al final del ciclo. La diferencia en las dos corrientes da lugar al voltamograma en forma de pico. Las condiciones experimentales típicas para la polarografía diferencial de pulso son\(\tau \approx 1 \text{ s}\), t p ≈ 50 ms,\(\Delta E_\text{p}\) ≈ 50 mV y\(\Delta E_\text{s}\) ≈ 2 mV.
El voltamograma para la polarografía diferencial de pulso es aproximadamente la primera derivada del voltamograma para la polarografía de pulso normal. Para ver por qué este es el caso, tenga en cuenta que el cambio de corriente sobre un cambio fijo en el potencial,\(\Delta i / \Delta E\), se aproxima a la pendiente del voltamograma para la polarografía de pulso normal. Puede recordar que la primera derivada de una función devuelve la pendiente de la función en cada punto. La primera derivada de una función sigmoidal es una función en forma de pico.
Otras formas de polarografía de pulso incluyen la polarografía de escalera (Figura 11.4.12 c) y la polarografía de onda cuadrada (Figura 11.4.12 d). Una ventaja de la polarografía de onda cuadrada es que podemos hacer\(\tau\) muy pequeñas, quizás tan pequeñas como 5 ms, en comparación con 1 s para otras formas de polarografía de pulso, lo que disminuye significativamente el tiempo de análisis. Por ejemplo, supongamos que necesitamos escanear un rango potencial de 400 mV. Si utilizamos polarografía de pulso normal con una\(\Delta E_\text{s}\) de 2 MV/ciclo y una\(\tau\) de 1 s/ciclo, entonces necesitamos 200 s para completar la exploración. Si utilizamos polarografía de onda cuadrada con una\(\Delta E_\text{s}\) de 2 MV/ciclo y una\(\tau\) de 5 ms/ciclo, podemos completar el escaneo en 1 s. A este ritmo, podemos adquirir un voltamograma completo usando una sola gota de Hg!
La polarografía se utiliza ampliamente para el análisis de iones metálicos y aniones inorgánicos, tales como\(\text{IO}_3^-\) y\(\text{NO}_3^-\). También podemos utilizar la polarografía para estudiar compuestos orgánicos con grupos funcionales fácilmente reducibles u oxidables, como carbonilos, ácidos carboxílicos y dobles enlaces carbono-carbono.
Voltamperometría hidrodinámica
En polarografía obtenemos una corriente limitante porque cada gota de mercurio mezcla la solución a medida que cae al fondo de la celda electroquímica. Si reemplazamos el DME por un electrodo sólido (ver Figura 11.4.3 ), aún podemos obtener una corriente limitante si agitamos mecánicamente la solución durante el análisis, usando una barra agitadora o girando el electrodo. A este enfoque lo llamamos voltamperometría hidrodinámica.
La voltametría hidrodinámica utiliza los mismos perfiles de potencial que en la polarografía, como una exploración lineal (Figura 11.4.11 ) o un pulso diferencial (Figura 11.4.12 b). Los voltammogramas resultantes son idénticos a los de la polarografía, excepto por la falta de oscilaciones de corriente por el crecimiento de las gotas de mercurio. Debido a que la voltametría hidrodinámica no se limita a electrodos de Hg, es útil para analitos que sufren oxidación o reducción a potenciales más positivos.
Voltamperometría
Otra técnica voltamétrica importante es la voltametría de decapado, que consiste en tres técnicas relacionadas: voltamperometría de extracción anódica, voltamperometría de extracción catódica y voltametría de extracción adsortiva. Debido a que la voltametría de decapado anódica es la más utilizada de estas técnicas, la consideraremos con mayor detalle.
La voltametría anódica de decapado consta de dos pasos (Figura 11.4.13 ). El primer paso es una electrólisis de potencial controlado en la que mantenemos el electrodo de trabajo, generalmente una gota de mercurio colgante o un electrodo de película de mercurio, a un potencial catódico suficiente para depositar el ion metálico en el electrodo. Por ejemplo, al analizar Cu 2 + la reacción de deposición es
\[\mathrm{Cu}^{2+}+2 e^{-} \rightleftharpoons \mathrm{Cu}(\mathrm{Hg}) \nonumber\]
donde Cu (Hg) indica que el cobre está amalgamado con el mercurio. Esta etapa sirve como medio para concentrar el analito transfiriéndolo del mayor volumen de la solución al menor volumen del electrodo. Durante la mayor parte de la electrólisis agitamos la solución para aumentar la velocidad de deposición. Cerca del final del tiempo de deposición, detenemos la agitación, eliminando la convección como modo de transporte de masa, y permitimos que la solución se vuelva quiescente. Los tiempos de deposición típicos de 1—30 min son comunes, con analitos a concentraciones más bajas que requieren tiempos más largos.
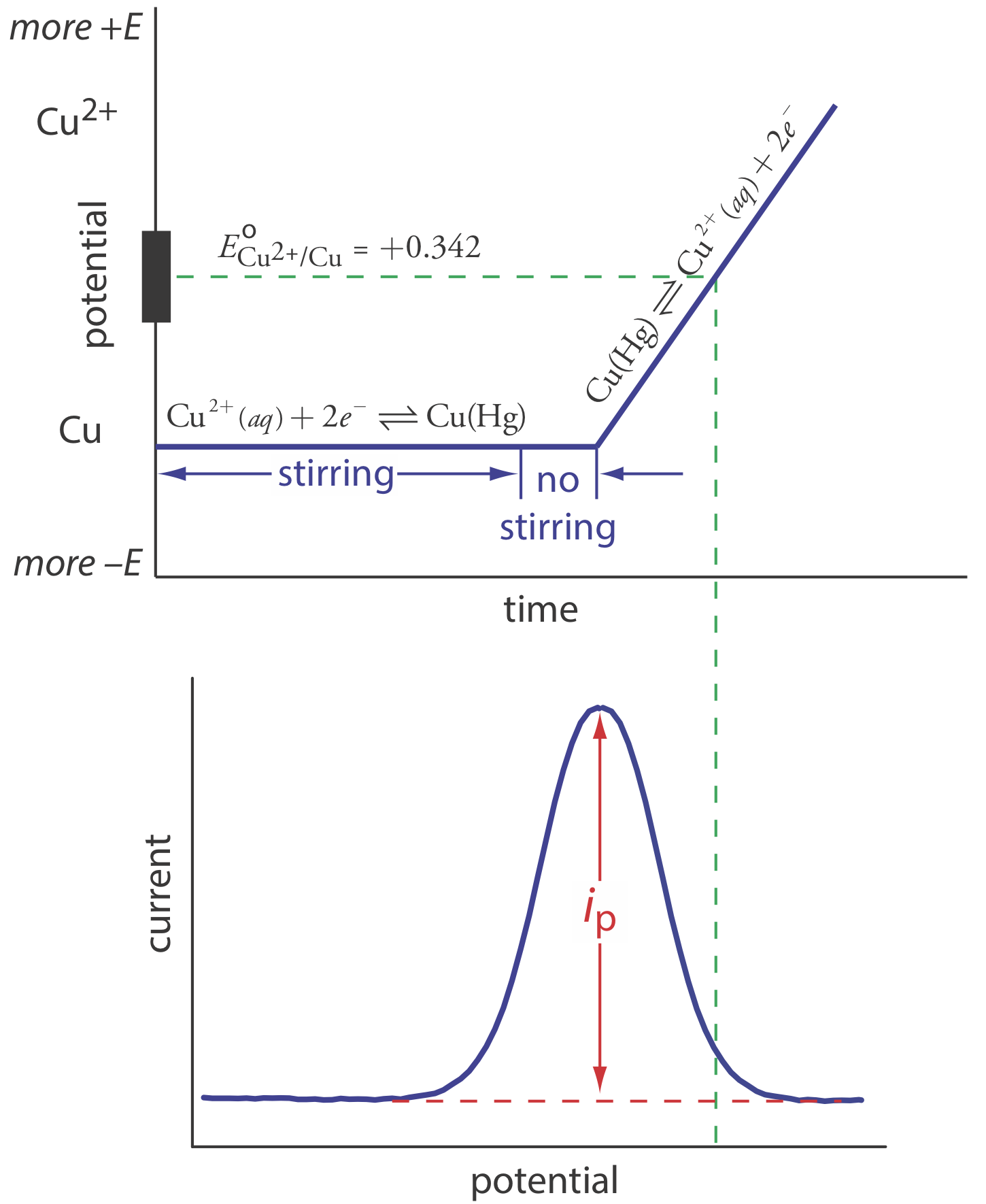
En el segundo paso, exploramos el potencial anódicamente, es decir, hacia un potencial más positivo. Cuando el potencial del electrodo de trabajo es suficientemente positivo, el analito es despojado del electrodo, volviendo a la solución en su forma oxidada.
\[\mathrm{Cu}(\mathrm{Hg})\rightleftharpoons \text{ Cu}^{2+}+2 e^{-} \nonumber\]
El monitoreo de la corriente durante la etapa de extracción da un voltamograma en forma de pico, como se muestra en la Figura 11.4.13 . La corriente pico es proporcional a la concentración del analito en la solución. Debido a que estamos concentrando el analito en el electrodo, los límites de detección son mucho más pequeños que otras técnicas electroquímicas. Una mejora de tres órdenes de magnitud, el equivalente a partes por mil millones en lugar de partes por millón, es rutinaria.
La voltametría anódica de decapado es muy sensible a las condiciones experimentales, las cuales debemos controlar cuidadosamente para obtener resultados precisos y precisos. Las variables clave incluyen el área de la película de mercurio o el tamaño de la gota de Hg colgante, el tiempo de deposición, el tiempo de reposo, la velocidad de agitación y la velocidad de barrido durante la etapa de extracción. La voltametría anódica de decapado es particularmente útil para metales que forman amalgamas con mercurio, varios ejemplos de los cuales se enumeran en la Tabla 11.4.1 .
El diseño experimental para la voltametría catódica de decapado es similar a la voltametría de extracción anódica con dos excepciones. Primero, la etapa de deposición implica la oxidación del electrodo de Hg\(\text{Hg}_2^{2+}\), que luego reacciona con el analito para formar una película insoluble en la superficie del electrodo. Por ejemplo, cuando Cl - es el analito, la etapa de deposición es
\[2 \mathrm{Hg}(l)+2 \mathrm{Cl}^{-}(a q) \rightleftharpoons \text{ Hg}_{2} \mathrm{Cl}_{2}(s)+2 e^{-} \nonumber\]
En segundo lugar, la extracción se realiza escaneando catódicamente hacia un potencial más negativo, reduciendo de\(\text{Hg}_2^{2+}\) nuevo a Hg y devolviendo el analito a la solución.
\[\mathrm{Hg}_{2} \mathrm{Cl}_{2}(s)+2 e^{-}\rightleftharpoons 2 \mathrm{Hg}( l)+2 \mathrm{Cl}^{-}(a q) \nonumber\]
En la Tabla 11.4.1 se enumeran varios analitos analizados exitosamente mediante voltamperometría de extracción catódica.
En la voltametría de extracción adsortiva, la etapa de deposición se produce sin electrólisis. En cambio, el analito se adsorbe a la superficie del electrodo. Durante la deposición mantenemos el electrodo a un potencial que potencia la adsorción. Por ejemplo, podemos adsorber una molécula neutra en una caída de Hg si aplicamos un potencial de —0.4 V versus el SCE, un potencial donde la carga superficial de mercurio es aproximadamente cero. Cuando la deposición está completa, exploramos el potencial en una dirección anódica o catódica, dependiendo de si estamos oxidando o reduciendo el analito. En la Tabla {{template.index (ID:1)} también se enumeran ejemplos de compuestos que han sido analizados por voltametría de extracción absortiva.
Voltamperometría cíclica
En las técnicas voltamétricas consideramos a este punto exploramos el potencial en una dirección, ya sea a potenciales más positivos o a potenciales más negativos. En voltamperometría cíclica completamos una exploración en ambas direcciones. La Figura 11.4.14 a muestra una señal típica de excitación potencial. En este ejemplo, primero exploramos el potencial a valores más positivos, dando como resultado la siguiente reacción de oxidación para la especie R.
\[R \rightleftharpoons O+n e^{-} \nonumber\]
Cuando el potencial alcanza un potencial de conmutación predeterminado, invertimos la dirección de la exploración hacia potenciales más negativos. Debido a que generamos la especie O en la exploración hacia adelante, durante la exploración inversa se reduce de nuevo a R.
\[O+n e^{-} \rightleftharpoons R \nonumber\]
La voltametría cíclica se realiza en una solución sin agitar, la cual, como se muestra en la Figura 11.4.14 b, da como resultado corrientes pico en lugar de corrientes limitantes. El voltamograma tiene picos separados para la reacción de oxidación y para la reacción de reducción, cada uno caracterizado por un potencial pico y una corriente pico.
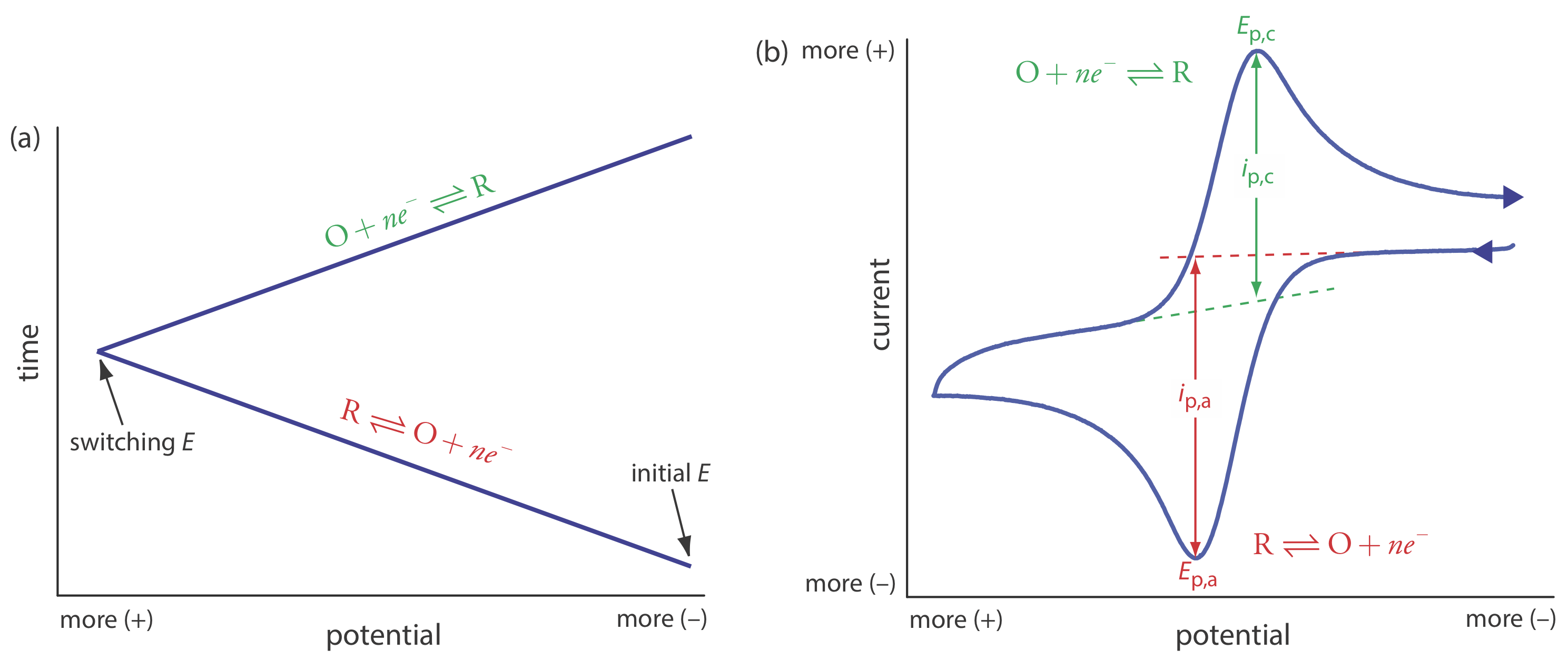
La corriente pico en voltamperometría cíclica viene dada por la ecuación de Randles-Sevcik
\[i_{p}=\left(2.69 \times 10^{5}\right) n^{3 / 2} A D^{1 / 2} \nu^{1 / 2} C_{A} \nonumber\]
donde n es el número de electrones en la reacción redox, A es el área del electrodo de trabajo, D es el coeficiente de difusión para las especies electroactivas,\(\nu\) es la velocidad de barrido, y C A es la concentración del especies electroactivas en el electrodo. Para un sistema de buen comportamiento, las corrientes pico anódica y catódica son iguales, y la relación i p, a/i p, c es 1.00. El potencial de media onda, E 1/2, está a medio camino entre los potenciales pico anódico y catódico.
\[E_{1 / 2}=\frac{E_{p, a}+E_{p, c}}{2} \nonumber\]
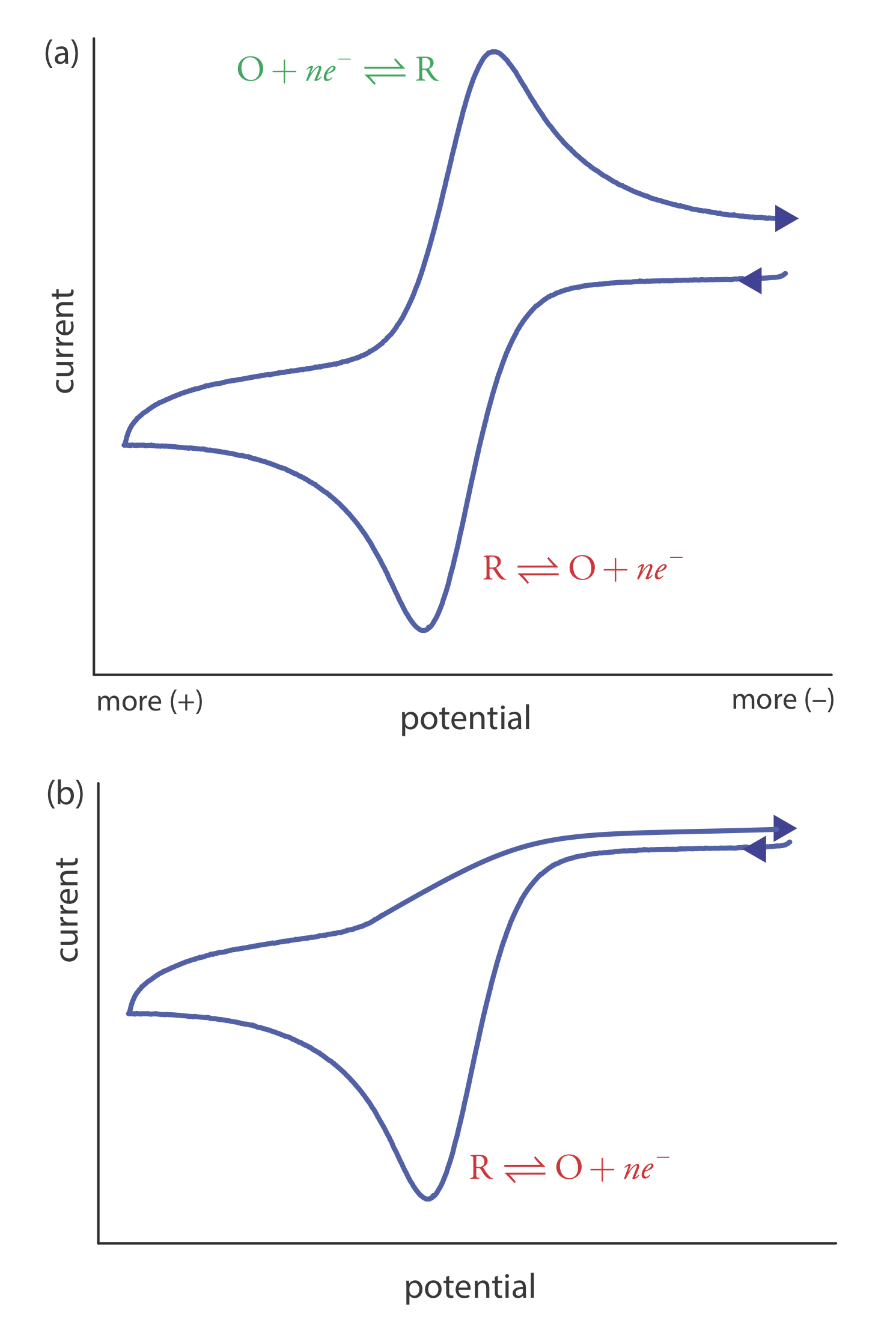
Escanear el potencial en ambas direcciones brinda la oportunidad de explorar el comportamiento electroquímico de las especies generadas en el electrodo. Esta es una clara ventaja de la voltametría cíclica sobre otras técnicas voltamétricas. La figura 11.4.15 muestra el voltamograma cíclico para el mismo par redox tanto a una velocidad de barrido más rápida como a otra más lenta. A la velocidad de escaneo más rápida, 11.4.15 a, vemos dos picos. A la velocidad de exploración más lenta en la Figura 11.4.15 b, sin embargo, el pico en el escaneo inverso desaparece. Una explicación para esto es que los productos de la reducción de R en el barrido directo tienen tiempo suficiente para participar en una reacción química cuyos productos no son electroactivos.
Amperometría
La técnica voltamétrica final que consideraremos es la amperometría, en la que aplicamos un potencial constante al electrodo de trabajo y medimos la corriente en función del tiempo. Debido a que no variamos el potencial, la amperometría no resulta en un voltamograma.
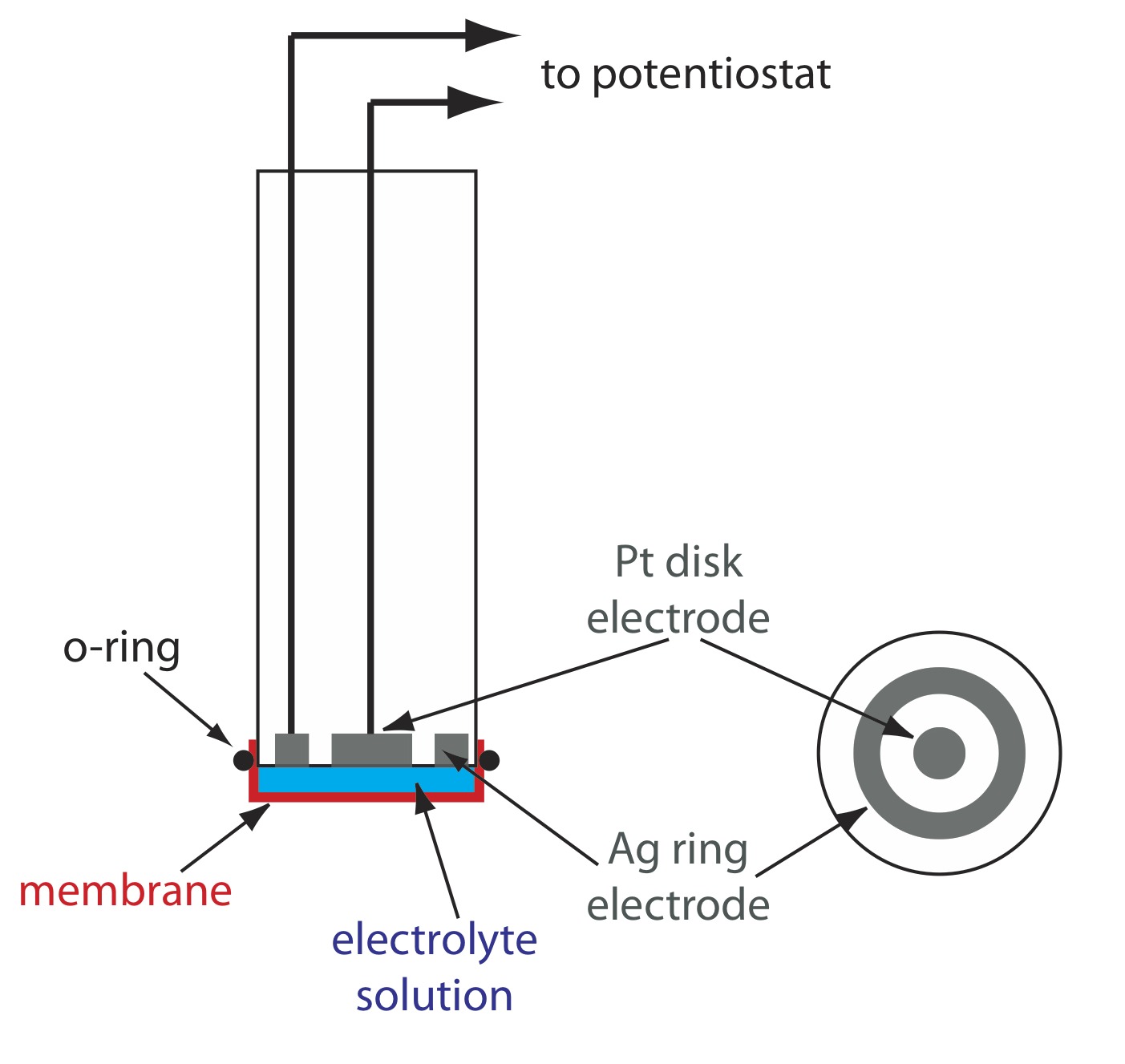
Una aplicación importante de la amperometría es en la construcción de sensores químicos. Uno de los primeros sensores amperométricos fue desarrollado en 1956 por L. C. Clark para medir O 2 disuelto en sangre. La Figura 11.4.16 muestra el diseño del sensor, el cual es similar a un electrodo de membrana potenciométrico. Una membrana delgada permeable a los gases se estira a través del extremo del sensor y se separa del electrodo de trabajo y del contraelectrodo por una solución delgada de KCl. El electrodo de trabajo es un cátodo de disco de Pt, y un ánodo de anillo de Ag sirve como contraelectrodo. Aunque varios gases pueden difundirse a través de la membrana, incluyendo O 2, N 2 y CO 2, solo el oxígeno sufre reducción en el cátodo
\[\mathrm{O}_{2}(g)+4 \mathrm{H}_{3} \mathrm{O}^{+}(a q)+4 e^{-}\rightleftharpoons 6 \mathrm{H}_{2} \mathrm{O}(l) \nonumber\]
con su concentración en la superficie del electrodo llegando rápidamente a cero. La concentración de O 2 en la superficie interna de la membrana se fija por su difusión a través de la membrana, lo que crea un perfil de difusión similar al de la Figura 11.4.8 . El resultado es una corriente de estado estacionario que es proporcional a la concentración de oxígeno disuelto. Debido a que el electrodo consume oxígeno, la muestra se agita para evitar el agotamiento de O 2 en la superficie exterior de la membrana.
La oxidación del ánodo de Ag es la otra semirreacción.
\[\mathrm{Ag}(s)+\text{ Cl}^{-}(a q)\rightleftharpoons \mathrm{AgCl}(s)+e^{-} \nonumber\]
Otro ejemplo de un sensor amperométrico es un sensor de glucosa. En este sensor se reemplaza la membrana única de la Figura 11.4.16 por tres membranas. La membrana más externa del policarbonato es permeable a la glucosa y al O 2. La segunda membrana contiene una preparación inmovilizada de glucosa oxidasa que cataliza la oxidación de la glucosa a gluconolactona y peróxido de hidrógeno.
\[\beta-\mathrm{D}-\text {glucose }(a q)+\text{ O}_{2}(a q)+\mathrm{H}_{2} \mathrm{O}(l)\rightleftharpoons \text {gluconolactone }(a q)+\text{ H}_{2} \mathrm{O}_{2}(a q) \nonumber\]
El peróxido de hidrógeno se difunde a través de la membrana más interna del acetato de celulosa donde experimenta oxidación en un ánodo de Pt.
\[\mathrm{H}_{2} \mathrm{O}_{2}(a q)+2 \mathrm{OH}^{-}(a q) \rightleftharpoons \text{ O}_{2}(a q)+2 \mathrm{H}_{2} \mathrm{O}(l)+2 e^{-} \nonumber\]
La Figura 11.4.17 resume las reacciones que se producen en este sensor amperométrico. El FAD es la forma oxidada del nucleótido de flavina adenina, el sitio activo de la enzima glucosa oxidasa, y FADH 2 es la forma reducida del sitio activo. Obsérvese que O 2 sirve como mediador, transportando electrones al electrodo.
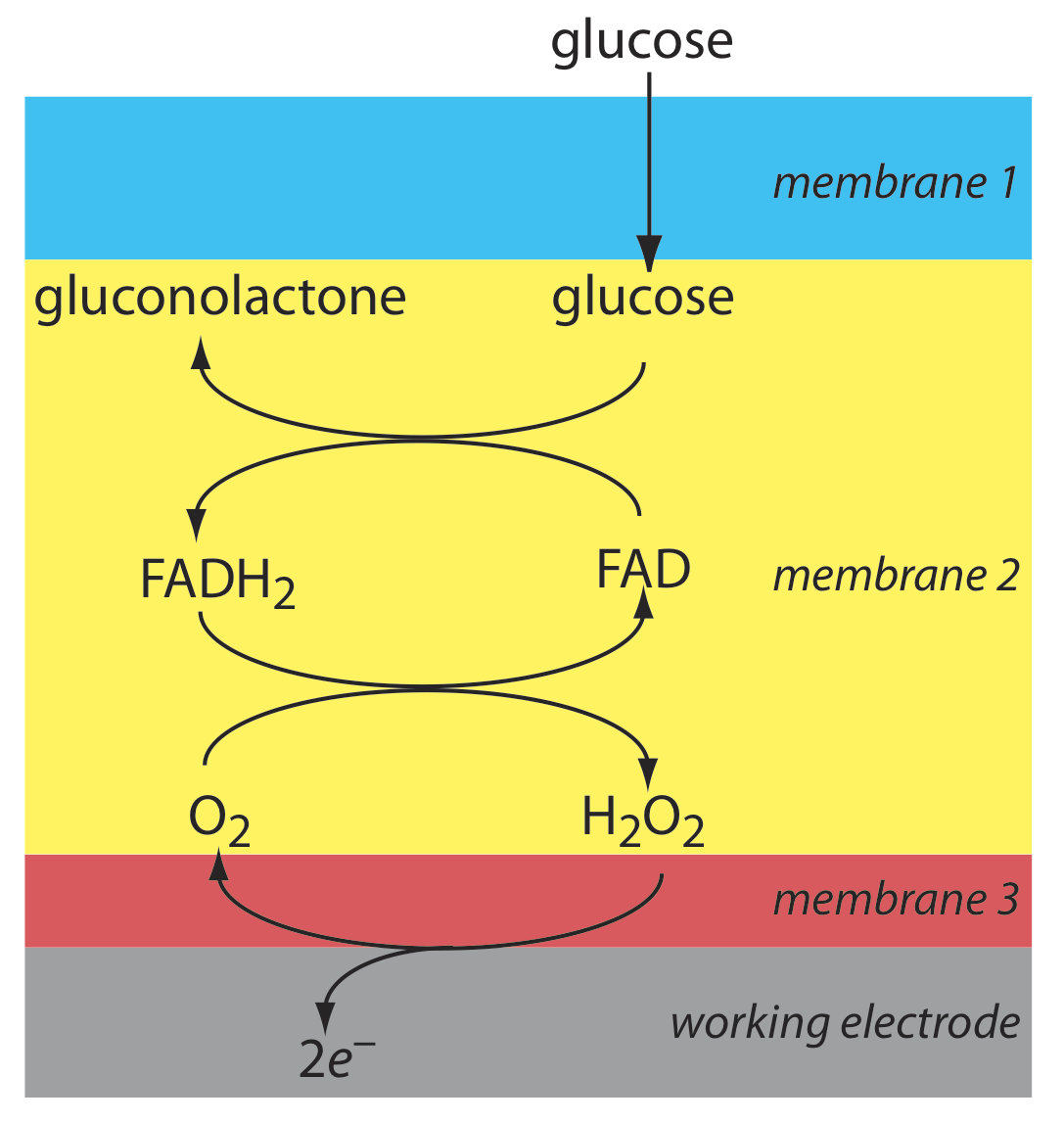
Al cambiar la enzima y el mediador, es fácil extender al sensor amperométrico en la Figura 11.4.17 al análisis de otros analitos. Por ejemplo, se ha desarrollado un sensor de CO 2 utilizando un sensor amperométrico O 2 con una membrana de dos capas, una de las cuales contiene una preparación inmovilizada de bacterias autotróficas [Karube, I.; Nomura, Y.; Arikawa, Y. Trends in Anal. Chem. 1995, 14, 295—299]. A medida que el CO 2 se difunde a través de las membranas, es convertido en O 2 por las bacterias, aumentando la concentración de O 2 en el cátodo de Pt.
Aplicaciones Cuantitativas
La voltametría se ha utilizado para el análisis cuantitativo de una amplia variedad de muestras, incluyendo muestras ambientales, muestras clínicas, formulaciones farmacéuticas, aceros, gasolina y aceite.
Selección de la Técnica Voltamétrica
La elección de qué técnica voltamétrica usar depende de las características de la muestra, incluyendo la concentración esperada del analito y la ubicación de la muestra. Por ejemplo, la amperometría es ideal para detectar analitos en sistemas de flujo, incluyendo el análisis in vivo de la sangre de un paciente o como sensor selectivo para el análisis rápido de un solo analito. La portabilidad de los sensores amperométricos, que son similares a los sensores potenciométricos, también los hacen ideales para estudios de campo. Aunque la voltametría cíclica se utiliza para determinar la concentración de un analito, otros métodos descritos en este capítulo son más adecuados para el trabajo cuantitativo.
La polarografía de pulso y la voltametría de pelado frecuentemente son intercambiables. La elección de qué técnica usar a menudo depende de la concentración del analito y de la precisión y precisión deseadas. Los límites de detección para la polarografía de pulso normal generalmente son del orden de 10 —6 M a 10 —7 M, y los de polarografía diferencial de pulso, escalera y polarografía de onda cuadrada están entre 10 —7 M y 10 —9 M. Debido a que concentramos el analito en voltamperometría de decapado, el límite de detección para muchos analitos es de tan solo 10 —10 M a 10 —12 M. Por otro lado, la corriente en la voltametría de extracción es mucho más sensible que la polarografía por pulsos a cambios en las condiciones experimentales, lo que puede conducir a una precisión y precisión más pobres . También podemos usar polarografía de pulso para analizar una gama más amplia de analitos inorgánicos y orgánicos porque no hay necesidad de depositar primero el analito en la superficie del electrodo.
La voltametría de decapado también sufre interferencias ocasionales cuando dos metales, como el Cu y el Zn, se combinan para formar un compuesto intermetálico en la amalgama de mercurio. El potencial de deposición de Zn. es suficientemente negativo para que cualquier Cu 2 + en la muestra también se deposite en la gota o película de mercurio, lo que lleva a la formación de compuestos intermetálicos como CuZn y CuZn 2. Durante la etapa de decapado, el zinc en los compuestos intermetálicos se tira a potenciales cercanos al del cobre, disminuyendo la corriente para el zinc a su potencial habitual y aumentando la corriente aparente para el cobre. Es posible superar este problema añadiendo un elemento que forme un compuesto intermetálico más fuerte con el metal interferente. Así, agregar Ga 3 + minimiza la interferencia de Cu al analizar Zn al formar un compuesto intermetálico de Cu y Ga.
Corrección de la Corriente Residual
En cualquier análisis cuantitativo debemos corregir la señal del analito para señales que surjan de otras fuentes. La corriente total, i tot, en voltamperometría consta de dos partes: la corriente de la oxidación o reducción del analito, i A, y una corriente de fondo o residual, i r.
\[i_{t o t}=i_{A}+i_{r} \nonumber\]
La corriente residual, a su vez, tiene dos fuentes. Una fuente es una corriente faradaica de la oxidación o reducción de los interferentes traza en la muestra, i int. La otra fuente es la corriente de carga, i ch, que acompaña un cambio en el potencial del electrodo de trabajo.
\[i_{r}=i_{\mathrm{int}}+i_{c h} \nonumber\]
Podemos minimizar la corriente faradaica debida a las impurezas preparando cuidadosamente la muestra. Por ejemplo, una impureza importante es el O 2 disuelto, que sufre una reducción en dos etapas: primero a H 2 O 2 a un potencial de —0.1 V versus el SCE, y luego a H 2 O a un potencial de —0.9 V contra el SCE. Eliminar O 2 disuelto burbujeando un gas inerte como N 2 a través de la muestra elimina esta interferencia. Después de retirar el O 2 disuelto, mantener una manta de N 2 sobre la parte superior de la solución evita que O 2 vuelva a entrar en la solución.
La celda de la Figura 11.4.4 muestra una línea típica de purga N 2.
Existen dos métodos para compensar la corriente residual. Un método consiste en medir la corriente total a potenciales donde la corriente faradaica del analito es cero y extrapolarla a otros potenciales. Este es el método que se muestra en la Figura 11.4.9 . Una ventaja de extrapolar es que no necesitamos adquirir datos adicionales. Una desventaja importante es que una extrapolación supone que cualquier cambio en la corriente residual con potencial es predecible, lo que puede no ser el caso. Un segundo enfoque, y más riguroso, es obtener un voltamograma para un blanco apropiado. La corriente residual del blanco se resta entonces de la corriente total de la muestra.
Análisis para componentes individuales
El análisis de una muestra con un solo analito es sencillo utilizando cualquiera de los métodos de estandarización discutidos en el Capítulo 5.
La concentración de As (III) en agua se determina mediante polarografía diferencial de pulso en HCl 1 M. El potencial inicial se establece en —0.1 V versus el SCE y se escanea hacia potenciales más negativos a una velocidad de 5 mV/s. La reducción de As (III) a As (0) ocurre a un potencial de aproximadamente —0.44 V frente al SCE. Las corrientes pico para un conjunto de soluciones estándar, corregidas para la corriente residual, se muestran en la siguiente tabla.
| [Como (III)] (µM) | i p (µM) |
|---|---|
| 1.00 | 0.298 |
| 3.00 | 0.947 |
| 6.00 | 1.83 |
| 9.00 | 2.72 |
¿Cuál es la concentración de As (III) en una muestra de agua si su pico de corriente es de 1.37 μA?
Solución
La regresión lineal da la curva de calibración mostrada en la Figura 11.4.18 , con una ecuación de
\[i_{p}=0.0176+3.01 \times[\mathrm{As}(\mathrm{III})] \nonumber\]
Sustituyendo la corriente pico de la muestra en la ecuación de regresión da la concentración de As (III) como 4.49 μM.

La concentración de cobre en una muestra de agua de mar se determina mediante voltametría de decapado anódico utilizando el método de adiciones estándar. El análisis de una muestra de 50.0-mL da una corriente pico de 0.886 μA. Después de agregar un pico de 5.00-μL de 10.0 mg/L Cu 2 +, la corriente pico aumenta a 2.52 μA. Calcular el μg/L de cobre en la muestra de agua de mar.
- Contestar
-
Para la voltametría de extracción anódica, la corriente pico, i p, es una función lineal de la concentración del analito
\[i_{p}=K \times C_{\mathrm{Cu}} \nonumber\]
donde K es una constante que da cuenta de parámetros experimentales como el área del electrodo, el coeficiente de difusión para Cu 2 +, el tiempo de deposición y la velocidad de agitación. Para el análisis de la muestra antes de la adición estándar sabemos que la corriente es
\[i_{p}=0.886 \ \mu \mathrm{A}=K \times C_{\mathrm{Cu}} \nonumber\]
y después de la adición estándar la corriente es
\[i_{p}=2.52 \ \mu \mathrm{A}=K\left\{C_{\mathrm{Cu}} \times \frac{50.00 \ \mathrm{mL}}{50.005 \ \mathrm{mL}}+\frac{10.00 \mathrm{mg} \mathrm{Cu}}{\mathrm{L}} \times \frac{0.005 \ \mathrm{mL}}{50.005 \ \mathrm{mL}}\right\} \nonumber\]
donde 50.005 mL es el volumen total después de agregar el pico de 5.00 μL. Resolver cada ecuación para K y combinar nos deja con la siguiente ecuación.
\[\frac{0.886 \ \mu \mathrm{A}}{C_{\mathrm{Cu}}}=K=\frac{2.52 \ \mu \mathrm{A}}{C_{\mathrm{Cu}} \times \frac{50.00 \ \mathrm{mL}}{50.005 \ \mathrm{mL}}+\frac{10.00 \ \mathrm{mg} \text{ Cu}}{\mathrm{L}} \times \frac{0.005 \ \mathrm{mL}}{50.005 \ \mathrm{mL}}} \nonumber\]
Resolver esta ecuación para C Cu da su valor como\(5.42 \times 10^{-4}\) mg Cu 2 + /L, o 0.542 μg Cu 2 + /L.
Análisis multicomponente
La voltametría es una técnica particularmente atractiva para el análisis de muestras que contienen dos o más analitos. Siempre que los analitos se comporten de forma independiente, el voltamograma de una mezcla multicomponente es una suma de los voltamogramas individuales de cada analito. Como se muestra en la Figura 11.4.19 , si la separación entre los potenciales de media onda o entre los potenciales pico es suficiente, podemos determinar la presencia de cada analito como si fuera el único analito en la muestra. La separación mínima entre los potenciales de media onda o los potenciales de pico para dos analitos depende de varios factores, incluyendo el tipo de electrodo y la señal de excitación potencial. Para la polarografía normal la separación es de al menos ±0.2—0.3 V, y la voltametría diferencial de pulso requiere una separación mínima de ±0.04—0.05 V.
Si los voltamogramas para dos analitos no están suficientemente separados, puede ser posible un análisis simultáneo. Un ejemplo de este enfoque se esboza el siguiente ejemplo.
El análisis polarográfico de pulso diferencial de una mezcla de indio y cadmio en HCl 0.1 M se complica por el solapamiento de sus respectivos voltamogramas [Lanza P. J. Chem. Educ. 1990, 67, 704—705]. El potencial pico para indio está en —0.557 V y el de cadmio está en —0.597 V. Cuando se analiza un estándar de indio de 0.800 ppm,\(\Delta i_p\) (en unidades arbitrarias) es 200.5 a —0.557 V y 87.5 a —0.597 V en relación con un electorde de referencia Ag/AgCl saturado. Una solución estándar de 0.793 ppm de cadmio tiene un\(\Delta i_p\) de 58.5 a —0.557 V y 128.5 a —0.597 V. Cuál es la concentración de indio y cadmio en una muestra si\(\Delta i_p\) es 167.0 a un potencial de —0.557 V y 99.5 a un potencial de —0.597V.
Solución
El cambio en la corriente,\(\Delta i_p\), en la polarografía diferencial de pulso es una función lineal de la concentración del analito
\[\Delta i_{p}=k_{A} C_{A} \nonumber\]
donde k A es una constante que depende del analito y del potencial aplicado, y C A es la concentración del analito. Para determinar las concentraciones de indio y cadmio en la muestra primero debemos encontrar el valor de k A para cada analito en cada potencial. Por simplicidad identificaremos el potencial de —0.557 V como E 1, y el de —0.597 V como E 2. Los valores de k A son
\[\begin{aligned} k_{\mathrm{In}, E_{1}} &=\frac{200.5}{0.800 \ \mathrm{ppm}}=250.6 \ \mathrm{ppm}^{-1} \\ k_{\mathrm{In}, E_{2}} &=\frac{87.5}{0.800 \ \mathrm{ppm}}=109.4 \ \mathrm{ppm}^{-1} \\ k_{\mathrm{Cd} E_{1}} &=\frac{58.5}{0.793 \ \mathrm{ppm}}=73.8 \ \mathrm{ppm}^{-1} \\ k_{\mathrm{Cd} E_{2}} &=\frac{128.5}{0.793 \ \mathrm{ppm}}=162.0 \ \mathrm{ppm}^{-1} \end{aligned} \nonumber\]
A continuación, escribimos ecuaciones simultáneas para la corriente en los dos potenciales.
\[\begin{array}{l}{\Delta i_{E_{1}}=167.0=250.6 \ \mathrm{ppm}^{-1} \times C_{\mathrm{In}}+73.8 \ \mathrm{ppm}^{-1} \times C_{\mathrm{Cd}}} \\ {\triangle i_{E_{2}}=99.5=109.4 \ \mathrm{ppm}^{-1} \times C_{\mathrm{In}}+162.0 \ \mathrm{ppm}^{-1} \times C_{\mathrm{Cd}}}\end{array} \nonumber\]
Resolviendo las ecuaciones simultáneas, que se deja como ejercicio, da la concentración de indio como 0.606 ppm y la concentración de cadmio como 0.205 ppm.
Muestras Ambientales
La voltametría es una de varias técnicas analíticas importantes para el análisis de metales traza en muestras ambientales, incluyendo agua subterránea, lagos, ríos y arroyos, agua de mar, lluvia y nieve. Los límites de detección en el nivel de partes por billón son rutinarios para muchos metales traza usando polarografía diferencial de pulso, con voltametría de extracción anódica que proporciona límites de detección de partes por billón para algunos metales traza.
Una aplicación ambiental interesante de la voltametría de extracción anódica es la determinación de la forma química de un metal traza dentro de una muestra de agua. La especiación es importante porque la biodisponibilidad, la toxicidad y la facilidad de transporte de un metal traza a menudo dependen de su forma química. Por ejemplo, un metal traza que está fuertemente unido a partículas coloidales generalmente no es tóxico porque no está disponible para formas de vida acuáticas. Desafortunadamente, la voltametría anódica de decapado no puede distinguir la forma química exacta de un metal traza porque especies estrechamente relacionadas, como Pb 2 + y PbCl +, producen un solo pico de extracción. En cambio, los metales traza se dividen en categorías “definidas operativamente” que tienen importancia ambiental.
Definido operacionalmente significa que un analito se divide en categorías por los métodos específicos utilizados para aislarlo de la muestra. Existen muchos ejemplos de definiciones operativas en la literatura ambiental. La distribución de los metales traza en suelos y sedimentos, por ejemplo, a menudo se define en términos de los reactivos utilizados para extraerlos; así, se puede encontrar una definición operativa para Zn 2 + en un sedimento lacustre como el extraído con acetato de sodio 1.0 M, o el extraído usando 1.0 M HCl.
Aunque existen muchos esquemas de especiación en la literatura ambiental, consideraremos uno propuesto por Batley y Florence [ver (a) Batley, G. E.; Florence, T. M. Anal. A lett. 1976, 9, 379—388; b) Batley, G. E.; Florence, T. M. Talanta 1977, 24, 151—158; c) Batley, G. E.; Florence, T. M. Anal. Chem. 1980, 52, 1962-1963; d) Florence, T. M., Batley, G. E.; CRC Crit. Rev. Anal. Chem. 1980, 9, 219—296]. Este esquema, que se describe en la Tabla 11.4.2 , combina la voltametría de extracción anódica con intercambio iónico e irradiación UV, dividiendo los metales traza solubles en siete grupos. En la primera etapa, la voltamperometría de extracción anódica en un tampón de ácido acético pH 4.8 diferencia entre metales lábiles y metales no lábiles. Solo los metales lábiles, aquellos presentes como iones hidratados, complejos débilmente unidos o débilmente adsorbidos sobre superficies coloidales, se depositan en el electrodo y dan lugar a una señal. La concentración total de metales se determina por ASV después de digerir la muestra en HNO 3 2 M durante 5 min, lo que convierte todos los metales en una forma lábil a ASV.
Una resina de intercambio iónico Chelex-100 diferencia aún más entre metales fuertemente unidos, generalmente metales unidos a sólidos inorgánicos y orgánicos, pero también aquellos fuertemente unidos a ligandos quelantes, y metales unidos más flojamente. Finalmente, la radiación UV diferencia entre metales unidos a fases orgánicas y fases inorgánicas. El análisis de muestras de agua de mar, por ejemplo, sugiere que el cadmio, el cobre y el plomo están presentes principalmente como complejos orgánicos lábiles o como adsorbatos lábiles en coloides orgánicos (Grupo II en la Tabla 11.4.2 ).
La polarografía diferencial de pulsos y la voltametría de decapado se utilizan para determinar trazas de metales en partículas transportadas por el aire, cenizas volantes incineradoras, rocas, minerales y sedimentos. Los metales traza, por supuesto, primero se ponen en solución usando una digestión o una extracción.
Los sensores amperométricos también se utilizan para analizar muestras ambientales. Por ejemplo, el sensor O 2 disuelto descrito anteriormente se utiliza para determinar el nivel de oxígeno disuelto y la demanda bioquímica de oxígeno, o DBO, de aguas y aguas residuales. Esta última prueba, que es una medida de la cantidad de oxígeno que requieren las bacterias acuáticas a medida que descomponen la materia orgánica, es importante a la hora de evaluar la eficiencia de una planta de tratamiento de aguas residuales y para monitorear la contaminación orgánica en aguas naturales. Un DBO alto sugiere que el agua tiene una alta concentración de materia orgánica. La descomposición de esta materia orgánica puede agotar seriamente el nivel de oxígeno disuelto en el agua, afectando negativamente a la vida acuática. Otros sensores amperométricos están disponibles para monitorear tensioactivos aniónicos en agua, y CO 2, H 2 SO 4 y NH 3 en gases atmosféricos.
Muestras Clínicas
La polarografía diferencial de pulso y la voltametría de extracción se utilizan para determinar la concentración de metales traza en una variedad de muestras clínicas, incluyendo sangre, orina y tejido. La determinación de plomo en la sangre es de considerable interés debido a las preocupaciones sobre el envenenamiento por plomo. Debido a que la concentración de plomo en la sangre es tan pequeña, la voltametría de extracción anódica frecuentemente es la técnica más apropiada. El análisis se complica, sin embargo, por la presencia de proteínas que pueden adsorberse al electrodo de mercurio, inhibiendo ya sea la deposición o el arrastre de plomo. Además, las proteínas pueden prevenir la electrodeposición de plomo a través de la formación de complejos estables y no lábiles. Digerir y macerar la muestra de sangre mini- empaña este problema. La polarografía diferencial de pulso es útil para el análisis cuantitativo rutinario de fármacos en fluidos biológicos, a concentraciones inferiores a 10 —6 M [Brooks, M. A. “Aplicación de la Electroquímica al Análisis Farmacéutico”, Capítulo 21 en Kissinger, P. T.; Heinemann, W. R., eds. Técnicas de Laboratorio en Química Electroanalítica, Marcel Dekker, Inc.: Nueva York, 1984, pp 539—568.]. Los sensores amperométricos que utilizan catalizadores enzimáticos también tienen muchos usos clínicos, varios ejemplos de los cuales se muestran en la Tabla 11.4.3 .
Muestras Diversas
Además de las muestras ambientales y clínicas, la polarografía diferencial de pulso y la voltametría de decapado se utilizan para el análisis de metales traza en otras muestras, incluyendo alimentos, aceros y otras aleaciones, gasolina, residuos de pólvora y productos farmacéuticos. La voltametría es una técnica importante para el análisis cuantitativo de compuestos orgánicos, particularmente en la industria farmacéutica donde se utiliza para determinar la concentración de fármacos y vitaminas en formulaciones. Por ejemplo, se dispone de métodos voltamétricos para el análisis cuantitativo de vitamina A, niacinamida y riboflavina. Cuando el compuesto de interés no es electroactivo, a menudo se puede derivatizar a una forma electroactiva. Un ejemplo es la determinación polarográfica de pulso diferencial de sulfanilamida, la cual se convierte en un colorante azo electroactivo por acoplamiento con ácido sulfámico y 1-naftol.
Método Representativo 11.4.1: Determinación de Cloropromazina en un Producto Farmacéutico
La mejor manera de apreciar los detalles teóricos y prácticos discutidos en esta sección es examinar cuidadosamente un método analítico típico. Aunque cada método es único, la siguiente descripción de la determinación de cloropromazina en un producto farmacéutico proporciona un ejemplo instructivo de un procedimiento típico. La descripción aquí se basa en un método de Pungor, E. A Practical Guide to Instrumental Analysis, CRC Press: Boca Raton, FL, 1995, pp. 34—37.
Descripción del método
La clorpromazina, también es conocida por su nombre comercial Thorazine, es un fármaco antipsicótico utilizado en el tratamiento de la esquizofrenia. La cantidad de clorpromazina en un producto farmacéutico se determina voltammétricamente en un electrodo de trabajo de grafito en una solución sin agitar, con calibración por el método de adiciones estándar.
Procedimiento
Agregar 10.00 mL de una solución de electrolito consistente en HCl 0.01 M y KCl 0.1 M a la celda electroquímica. Colocar un electrodo de trabajo de grafito, un electrodo auxiliar de Pt y un electrodo de referencia SCE en la celda, y registrar el voltamograma de 0.2 V a 2.0 V a una velocidad de barrido de 50 mV/s. Pesar una cantidad apropiada del producto farmacéutico y disolverlo en una pequeña cantidad del electrolito. Transfiera la solución a un matraz aforado de 100 ml y diluya al volumen con el electrolito. Filtrar una pequeña cantidad de la solución diluida y transferir 1.00 mL del filtrado a la celda voltamétrica. Mezclar el contenido de la celda voltamétrica y dejar que la solución se asiente durante 10 s antes de registrar el voltamograma. Devolver el potencial a 0.2 V, agregar 1.00 mL de un estándar de clorpromazina y registrar el voltamograma. Reporte el %w/w de clorpromazina en la formulación.
Preguntas
1. ¿La clorpromazina está sometida a oxidación o reducción en el electrodo de trabajo de grafito?
Debido a que estamos explorando hacia potenciales más positivos, estamos oxidando la clorpromazina.
2. ¿Por qué este procedimiento utiliza un electrodo de grafito en lugar de un electrodo de Hg?
Como se muestra en la Figura 11.4.2 , la ventana de potencial para un electrodo de Hg se extiende desde aproximadamente —0.3 V hasta entre —1V y —2 V, de- pendiente en el pH. Debido a que estamos escaneando el potencial de 0.2 V a 2.0 V, no podemos usar un electrodo de Hg.
3. Muchos procedimientos voltamétricos requieren que primero eliminemos O 2 disuelto burbujeando N 2 a través de la solución. ¿Por qué esto no es necesario para este análisis?
O 2 disuelto es un problema cuando exploramos hacia potenciales más negativos, ya que su reducción puede producir una corriente catódica significativa. En este procedimiento estamos explorando hacia potenciales más positivos y generando corrientes anódicas; así, O 2 disuelto no es un interferente y no necesita ser eliminado.
4. ¿Cuál es el propósito de registrar un voltamograma en ausencia de clorpromazina?
Este voltamograma sirve como una pieza en blanco, que proporciona una medición de la corriente residual debida al electrolito. Debido a que la ventana de potencial para un electrodo de trabajo de grafito (ver Figura 11.4.2 ) no se extiende a 2.0 V, existe una corriente residual anódica medible debido a la oxidación del solvente. Habiendo medido esta corriente residual, podemos restarla de la corriente total en presencia de clorpromazina.
5. Con base en la descripción de este procedimiento, cuál es la forma del voltamograma resultante. Es posible que desee revisar las tres formas comunes que se muestran en la Figura 11.4.9 .
Debido a que la solución no está agitada, el voltamograma tendrá una corriente pico similar a la que se muestra en la Figura 11.4.9 b.
Aplicaciones de Caracterización
En la sección anterior aprendimos a usar la voltametría para determinar la concentración de un analito en una variedad de muestras diferentes. También podemos utilizar la voltametría para caracterizar las propiedades de un analito, incluyendo verificar su reversibilidad electroquímica, determinar el número de electrones transferidos durante su oxidación o reducción, y determinar su constante de equilibrio en una reacción química acoplada.
Reversibilidad Electroquímica y Determinación de n
Anteriormente en este capítulo derivamos una relación entre E 1/2 y el potencial de estado estándar para una pareja redox (Ecuación\ ref {11.9}), señalando que una reacción redox debe ser electroquímicamente reversible. ¿Cómo podemos saber si una reacción redox es reversible observando su voltamograma? Para una reacción redox reversible, la Ecuación\ ref {11.8}, que repetimos aquí, describe la relación entre potencial y corriente para un experimento voltamétrico con una corriente limitante.
\[E=E_{O / R}^{\circ}-\frac{0.05916}{n} \log \frac{K_{O}}{K_{R}}-\frac{0.05916}{n} \log \frac{i}{i_{l} - i} \nonumber\]
Si una reacción es electroquímicamente reversible, una gráfica de E versus log (i/i l — i) es una línea recta con una pendiente de —0.05916/ n. Además, la pendiente debe dar un valor entero para n.
Los siguientes datos se obtuvieron de un voltamograma hidrodinámico de barrido lineal de una reacción de reducción reversible.
| E (V frente a SCE) | corriente (μA) |
|---|---|
| —0.358 | 0.37 |
| —0.372 | 0.95 |
| —0.382 | 1.71 |
| —0.400 | 3.48 |
| —0.410 | 4.20 |
| —0.435 | 4.97 |
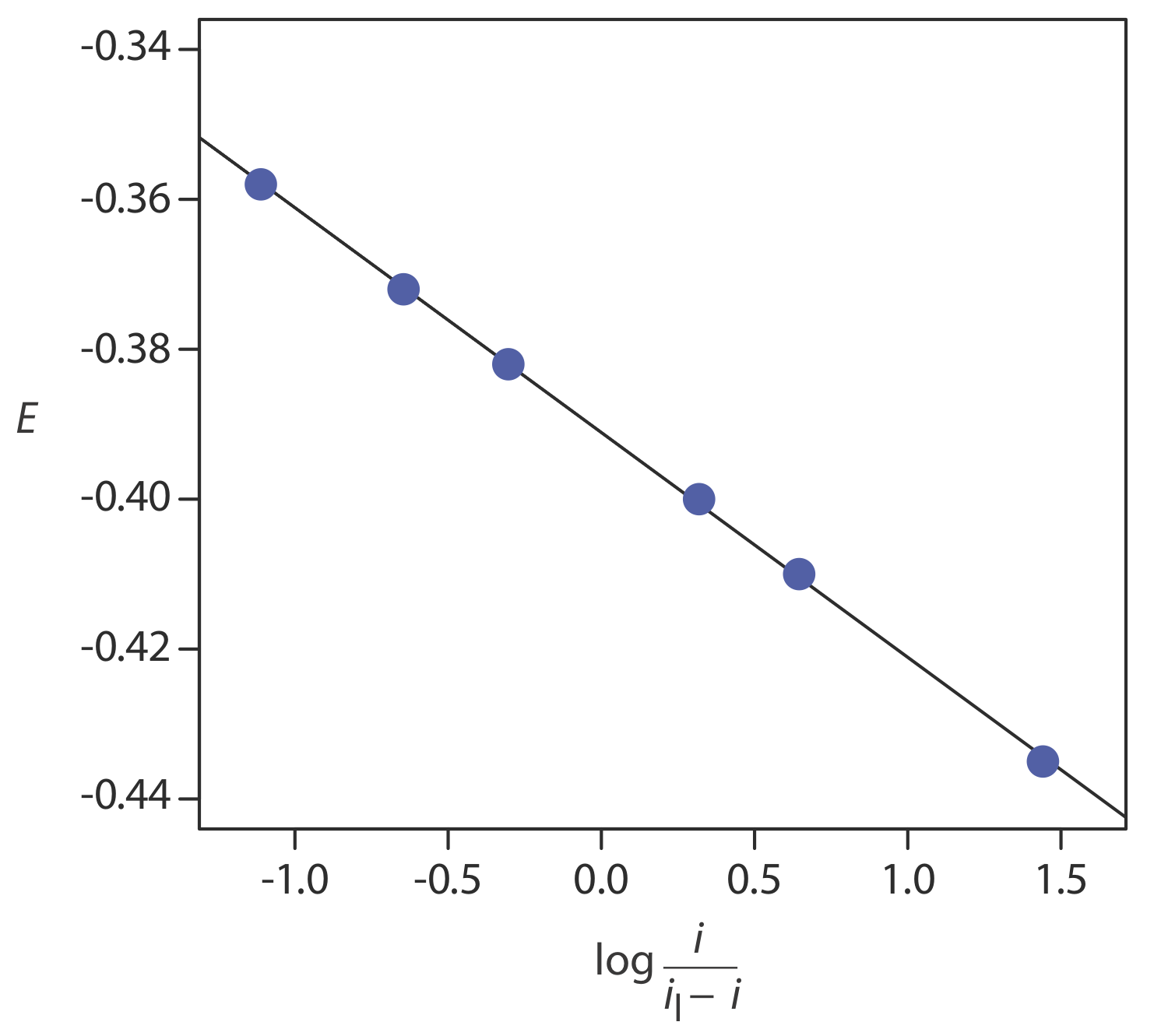
La corriente límite es de 5.15 μA. Mostrar que la reacción de reducción es reversible, y determinar valores para n y para E 1/2.
Solución
La figura 11.4.20 muestra una gráfica de E versus log (i/i l — i). Debido a que el resultado es una línea recta, sabemos que la reacción es electroquímicamente reversible bajo las condiciones del experimento. Un análisis de regresión lineal da la ecuación para la línea recta como
\[E=-0.391 \mathrm{V}-0.0300 \log \frac{i}{i_{l}-i} \nonumber\]
De la Ecuación\ ref {11.8}, la pendiente es equivalente a —0.05916/ n; resolviendo para n da un valor de 1.97, o 2 electrones. A partir de la Ecuación\ ref {11.8} y la Ecuación\ ref {11.9}, sabemos que E 1/2 es la intercepción y para una gráfica de E versus log (i/i l — i); así, E 1/2 para los datos en este ejemplo es —0.391 V versus el SCE.
También podemos utilizar voltamperometría cíclica para evaluar la reversibilidad electroquímica observando la diferencia entre los potenciales máximos para los escaneos anódicos y catódicos. Para una reacción electroquímicamente reversible, la siguiente ecuación es cierta.
\[\Delta E_{p}=E_{p, a}-E_{p, c}=\frac{0.05916 \ \mathrm{V}}{n} \nonumber\]
Como ejemplo, para una reducción de dos electrones esperamos una\(\Delta E_p\) de aproximadamente 29.6 mV. Para una reacción electroquímicamente irreversible el valor de\(\Delta E_p\) es mayor de lo esperado.
Determinación de constantes de equilibrio para reacciones químicas acopladas
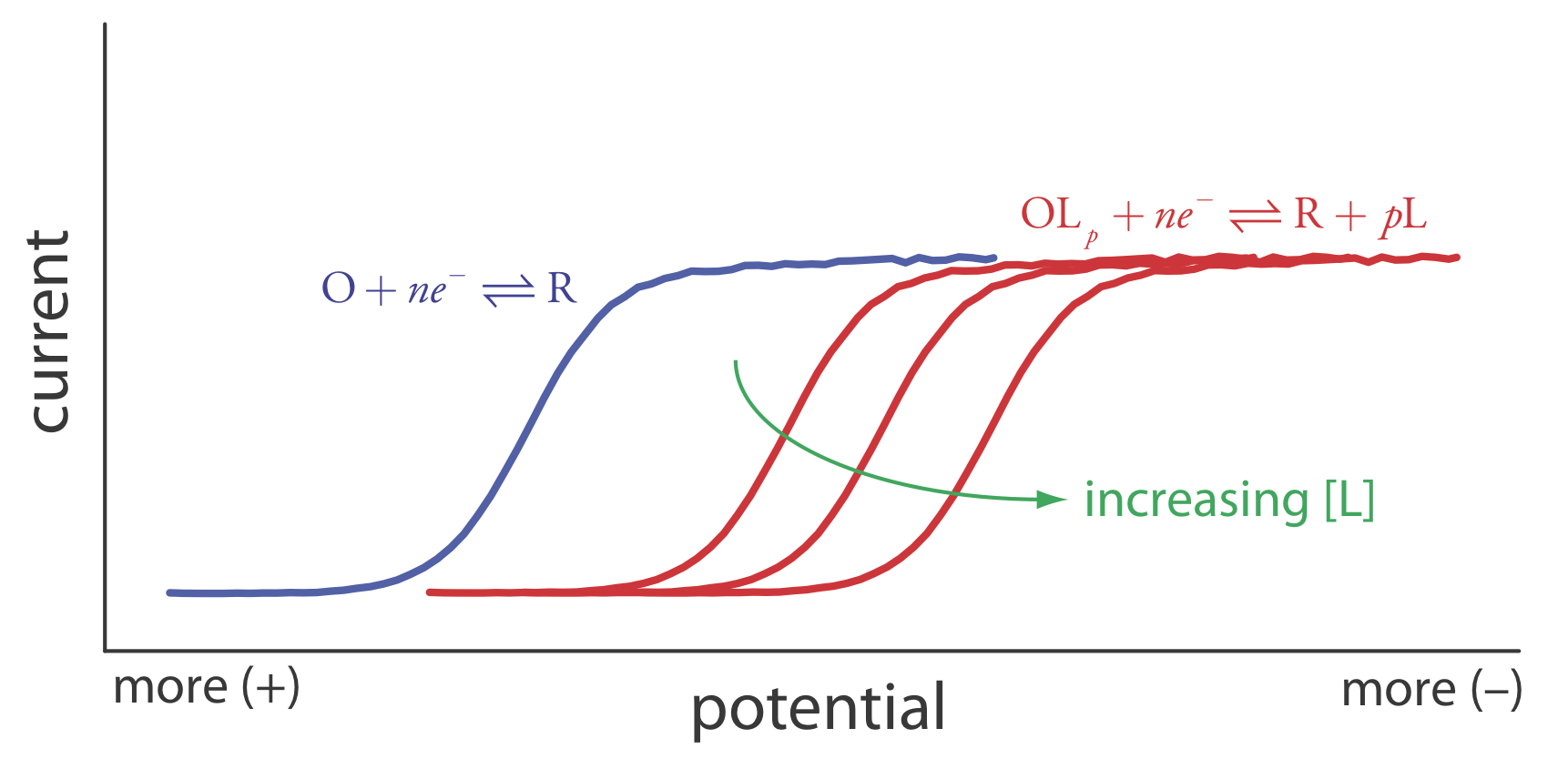
Otra aplicación importante de la voltametría es determinar la constante de equilibrio para una reacción en solución que se acopla a una reacción redox. La presencia de la reacción en solución afecta la facilidad de transferencia de electrones en la reacción redox, desplazando E 1/2 a un potencial más negativo o a un potencial más positivo. Consideremos, por ejemplo, la reducción de O a R
\[O+n e^{-} \rightleftharpoons R \nonumber\]
el voltamograma para el que se muestra en la Figura 11.4.21 . Si introducimos un ligando, L, que forma un complejo fuerte con O, entonces también debemos considerar la reacción
\[O+p L\rightleftharpoons O L_{p} \nonumber\]
En presencia del ligando, la reacción redox global es
\[O L_{p}+n e^{-} \rightleftharpoons R+p L \nonumber\]
Debido a su estabilidad, la reducción del complejo OL p es menos favorable que la reducción de O. Como se muestra en la Figura 11.4.21 , el voltamograma resultante se desplaza a un potencial que es más negativo que el de O. Además, el cambio en el voltamograma aumenta a medida que aumentamos la concentración del ligando.
Podemos usar este cambio en el valor de E 1/2 para determinar tanto la estequiometría como la constante de formación para un complejo metal-ligando. Para derivar una relación entre las variables relevantes comenzamos con dos ecuaciones: la ecuación de Nernst para la reducción de O
\[E=E_{O / R}^{\circ}-\frac{0.05916}{n} \log \frac{[R]_{x=0}}{[O]_{x=0}} \label{11.10}\]
y la constante de estabilidad,\(\beta_p\) para el complejo metal-ligando en la superficie del electrodo.
\[\beta_{p} = \frac{\left[O L_p\right]_{x = 0}}{[O]_{x = 0}[L]_{x = 0}^p} \label{11.11}\]
En ausencia de ligando el potencial de media onda ocurre cuando [R] x = 0 y [O] x = 0 son iguales; así, a partir de la ecuación de Nernst tenemos
\[\left(E_{1 / 2}\right)_{n c}=E_{O / R}^{\circ} \label{11.12}\]
donde el subíndice “nc” significa que el complejo no está presente.
Cuando el ligando está presente debemos dar cuenta de su efecto sobre la concentración de O. Resolviendo la Ecuación\ ref {11.1} para [O] x = 0 y sustituyendo en la Ecuación\ ref {11.10} da
Si la constante de formación es suficientemente grande, de tal manera que esencialmente todo O está presente como el complejo OL p, entonces [R] x = 0 y [OL p] x = 0 son igual al potencial de media onda, y la Ecuación\ ref {11.13} simplifica a
donde el subíndice “c” indica que el complejo está presente. Definir\(\Delta E_{1/2}\) como
\[\triangle E_{1 / 2}=\left(E_{1 / 2}\right)_{c}-\left(E_{1 / 2}\right)_{n c} \label{11.15}\]
y sustituyendo la Ecuación\ ref {11.12} y la Ecuación\ ref {11.14} y expandir el término logarítmico nos deja con la siguiente ecuación.
\[\Delta E_{1 / 2}=-\frac{0.05916}{n} \log \beta_{p}-\frac{0.05916 p}{n} \log {[L]} \label{11.16}\]
Una gráfica\(\Delta E_{1/2}\) versus log [L] es una línea recta, con una pendiente que es función del coeficiente estequiométrico del complejo metal-ligando, p, y una intersección y que es función de su constante de formación\(\beta_p\).
Un voltamograma para la reducción de dos electrones (n = 2) de un metal, M, tiene un potencial de media onda de —0.226 V versus el SCE. En presencia de un exceso de ligando, L, se registran los siguientes potenciales de media onda.
| [L] (M) | (E 1/2) c (V frente a SCE) |
|---|---|
| 0.020 | —0.494 |
| 0.040 | —0.512 |
| 0.060 | —0.523 |
| 0.080 | —0.530 |
| 0.100 | —0.536 |
Determinar la estequiometría del complejo metal-ligando y su constante de formación.
Solución
Comenzamos calculando valores de\(\Delta E_{1/2}\) usar la Ecuación\ ref {11.15}, obteniendo los valores en la siguiente tabla.
| [L] (M) | \(\Delta E_{1/2}\)(V vs. SCE) |
|---|---|
| 0.020 | \ (\ Delta E_ {1/2}\) (V vs SCE) ">—0.268 |
| 0.040 | \ (\ Delta E_ {1/2}\) (V vs SCE) ">—0.286 |
| 0.060 | \ (\ Delta E_ {1/2}\) (V vs SCE) ">—0.297 |
| 0.080 | \ (\ Delta E_ {1/2}\) (V vs SCE) ">—0.304 |
| 0.100 | \ (\ Delta E_ {1/2}\) (V vs SCE) ">—0.310 |
La figura 11.4.22 muestra la gráfica resultante de\(\Delta E_{1/2}\) como una función de log [L]. Un análisis de regresión lineal da la ecuación para la línea recta como
\[\triangle E_{1 / 2}=-0.370 \mathrm{V}-0.0601 \log {[L]} \nonumber\]
De la Ecuación\ ref {11.16} sabemos que la pendiente es igual a —0.05916 p/n. Usando la pendiente y n = 2, resolvemos para p obteniendo un valor de 2.03 ≈ 2. La estequiometría del complejo, por lo tanto, es ML 2. También sabemos, por la Ecuación\ ref {11.16}, que la intersección y es equivalente a — (0.05916/ n) log\(\beta_p\). Resolviendo para\(\beta_2\) da una constante de formación de\(3.2 \times 10^{12}\).
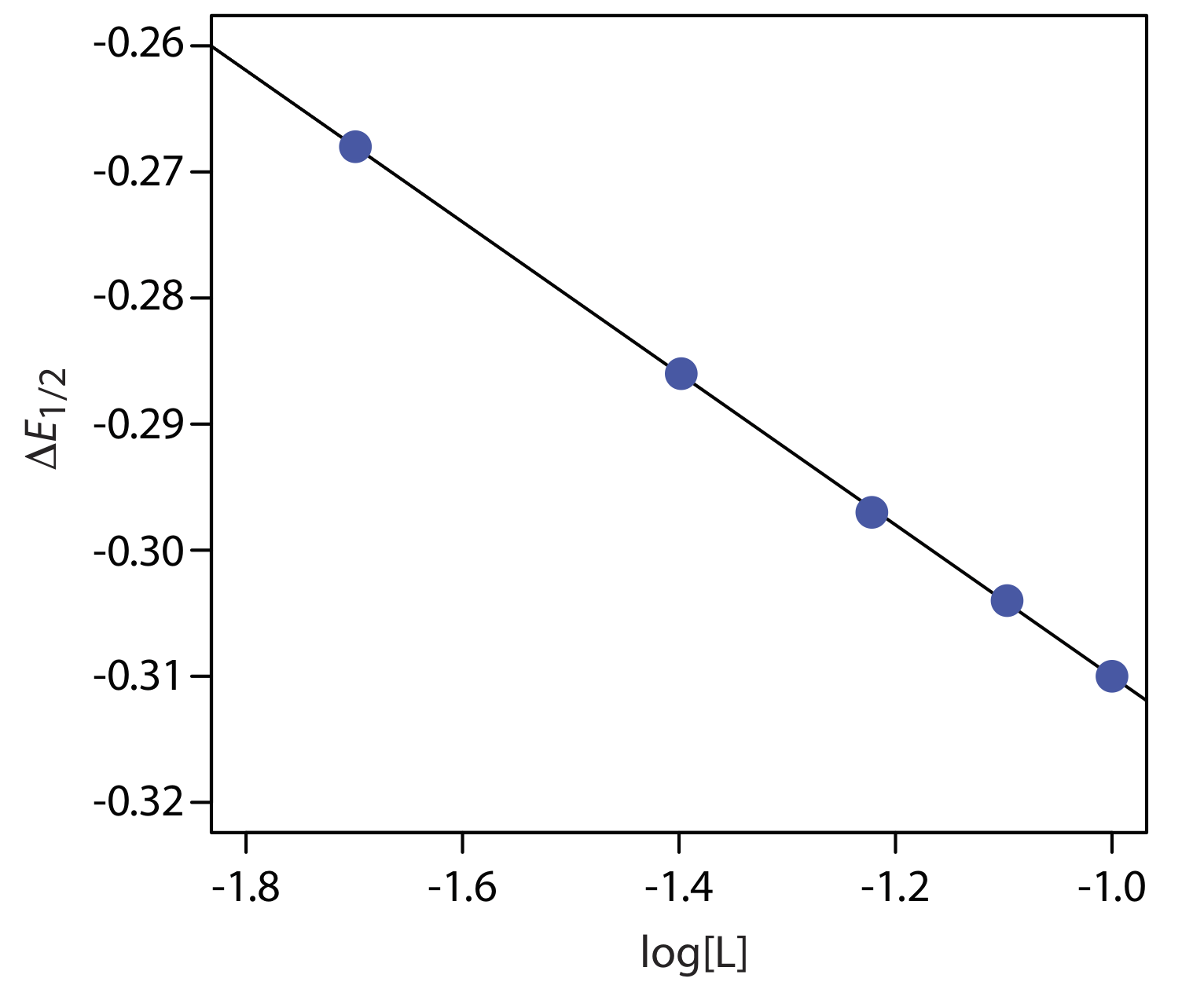
El voltamograma para Cd 2 + 0.50 mM tiene un E 1/2 de —0.565 V versus un SCE. Después de hacer la solución 0.115 M en etilendiamina, E 1/2 es —0.845 V, y E 1/2 es —0.873 V cuando la solución es 0.231 M en etilendiamina. Determinar la estequiometría del complejo Cd 2 + —etilendiamina y su constante de formación. Los datos en este problema provienen de Morinaga, K. “Estudios Polarográficos de Complejos Metálicos. V. Complejos de Etilendiamina de Cadmio, Níquel y Zinc”, Bol. Chem. Soc. Japón 1956, 29, 793—799.
- Contestar
-
Para simplificar, usaremos en como notación taquigráfica para etilendiamina. De los tres potenciales de media onda tenemos un\(\Delta E_{1/2}\) de —0.280 V para 0.115 M en y a\(\Delta E_{1/2}\) de —0.308 V para 0.231 M en. Usando la ecuación\ ref {11.16} escribimos las siguientes dos ecuaciones.
\[\begin{array}{l}{-0.280=-\frac{0.05916}{2} \log \beta_{p}-\frac{0.05916 p}{2} \log (0.115)} \\ {-0.308=-\frac{0.05916}{2} \log \beta_{p}-\frac{0.05916 p}{2} \log (0.231)}\end{array} \nonumber\]
Para resolver el valor de p, primero restamos la segunda ecuación de la primera ecuación
\[0.028=-\frac{0.05916 p}{2} \log (0.115)-\left\{-\frac{0.05916 p}{2} \log (0.231)\right\} \nonumber\]
lo que elimina el término con\(\beta_p\). A continuación resolvemos esta ecuación para p
\[0.028=\left(2.778 \times 10^{-2}\right) \times p-\left(1.882 \times 10^{-2}\right) \times p =\left(8.96 \times 10^{-3}\right) \times p \nonumber\]
obteniendo un valor de 3.1, o p ≈ 3. Así, el complejo es Cd (en) 3. Para encontrar el complejo formativo,\(\beta_3\), volvemos a la Ecuación\ ref {11.16}, usando nuestro valor para p. Usando los datos para una concentración en de 0.115 M
\[\begin{aligned}-0.280=-& \frac{0.05916}{2} \log \beta_{3}-\frac{0.05916 \times 3}{2} \log (0.115) \\ &-0.363=-\frac{0.05916}{2} \log \beta_{3} \end{aligned} \nonumber\]
da un valor para\(\beta_3\) de\(1.92 \times 10^{12}\). El uso de los datos para una concentración en de 0.231 M da un valor de\(2.10 \times 10^{12}\).
Como sugiere la Figura 11.4.15 , la voltametría cíclica es una de las técnicas electroquímicas más potentes para explorar el mecanismo de las reacciones electroquímicas y químicas acopladas. El tratamiento de este aspecto de la voltametría cíclica está más allá del nivel de este texto, aunque puede consultar los recursos adicionales de este capítulo para obtener información adicional.
Evaluación
Escala de Operación
Los niveles de detección en el nivel de partes por millón son rutinarios. Para algunos analitos y para algunas técnicas voltamétricas, son posibles menores límites de detección. Los límites de detección a partes por mil millones y a nivel de parte por billón son posibles con voltametría de extracción. Aunque la mayoría de los análisis se realizan en celdas electroquímicas convencionales utilizando macromuestras, la disponibilidad de microelectrodos con diámetros tan pequeños como 2 μm, permite el análisis de muestras con volúmenes inferiores a 50 μL. Por ejemplo, la concentración de glucosa en neuronas de caracol de estanque de 200 μm se monitoreó exitosamente usando un electrodo amperométrico de glucosa con una punta de 2 mm [Abe, T.; Lauw, L. L.; Ewing, A. G. J. Am. Chem. Soc. 1991, 113, 7421—7423].
Precisión
La precisión de un análisis voltamétrico generalmente está limitada por nuestra capacidad de corregir las corrientes residuales, particularmente las debidas a la carga. Para un analito en el nivel de partes por millón, una precisión de ±1— 3% es rutinaria. La precisión disminuye para muestras con concentraciones significativamente menores de analito.
Precisión
La precisión generalmente está limitada por la incertidumbre en la medición de la corriente límite o la corriente de pico. En la mayoría de las condiciones, una precisión de ±1— 3% es razonable. Una excepción es el análisis de analitos de ultratraza en matrices complejas por voltametría de decapado, en el que la precisión puede ser tan pobre como ± 25%.
Sensibilidad
En muchos experimentos voltamétricos, podemos mejorar la sensibilidad ajustando las condiciones experimentales. Por ejemplo, en la voltametría de decapado podemos mejorar la sensibilidad aumentando el tiempo de deposición, aumentando la velocidad de barrido de potencial lineal, o mediante el uso de una técnica de pulso diferencial. Una razón por la que las técnicas de pulso potenciales son populares es que proporcionan una mejora en la corriente en relación con una exploración de potencial lineal.
Selectividad
La selectividad en voltamperometría está determinada por la diferencia entre los potenciales de media onda o los potenciales de pico, con una diferencia mínima de ±0.2—0.3 V para una exploración de potencial lineal y ±0.04—0.05 V para voltamperometría diferencial de pulsos. A menudo podemos mejorar la selectividad ajustando las condiciones de la solución. La adición de un ligando complejante, por ejemplo, puede desplazar sustancialmente el potencial donde una especie se oxida o se reduce a un potencial donde ya no interfiere con la determinación de un analito. Otros parámetros de solución, como el pH, también se pueden usar para mejorar la selectividad.
Tiempo, Costo y Equipo
La instrumentación comercial para voltamperometría varía desde <$1000 para instrumentos simples hasta >$20,000 para un instrumento más sofisticado. En general, la instrumentación menos costosa se limita a las exploraciones de potencial lineal. Los instrumentos más caros proporcionan señales de excitación de potencial más complejas usando pulsos potenciales. A excepción de la voltametría de decapado, que necesita un largo tiempo de deposición, los análisis voltamétricos son relativamente rápidos.