
11.5: Problemas
- Page ID
- 75820

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)1. Identificar el ánodo y el cátodo para las siguientes celdas electroquímicas, e identificar la reacción de oxidación o reducción en cada electrodo.
(a) Pt| FeCl 2 (aq, 0.015), FeCl 3 (aq, 0.045) || AgNo 3 (aq, 0.1) | Ag
(b) Ag | AgBr (s), NaBr (aq, 1.0) || CdCl 2 (aq, 0.05) | Cd
(c) Pb | PbSO 4 (s), H 2 SO 4 (aq, 1.5) || H 2 SO 4 (aq, 2.0), PbSO 4 (s) | PbO 2
2. Calcular el potencial para cada celda electroquímica en el problema 1. Los valores entre paréntesis son las actividades de las especies asociadas.
3. Calcular la actividad de KI, x, en la siguiente celda electroquímica si el potencial es +0.294 V.
Ag | AgCl (s), NaCl (aq, 0.1) || KI (aq, x), I 2 (s) | Pt
4. ¿Qué reacción nos impide usar Zn como electrodo de primer tipo en una solución ácida? ¿Qué otros metales espera que se comporten de la misma manera que el Zn cuando se sumerge en una solución ácida?
5. Creager y sus colegas diseñaron un electrodo selectivo de iones salicilato usando una membrana de PVC impregnada con salicilato de tetraalquilamonio [Creager, S. E.; Lawrence, K. D.; Tibbets, C. R. J. Chem. Educ. 1995, 72, 274—276]. Para determinar el coeficiente de selectividad del electrodo selectivo de iones para el benzoato, prepararon un conjunto de patrones de calibración de salicilato en los que la concentración de benzoato se mantuvo constante a 0.10 M. Utilizando los siguientes datos, determinar el valor del coeficiente de selectividad.
| [salicilato] (M) | potencial (mV) |
|---|---|
| 1.0 | 20.2 |
| \(1.0 \times 10^{-1}\) | 73.5 |
| \(1.0 \times 10^{-2}\) | 126 |
| \(1.0 \times 10^{-3}\) | 168 |
| \(1.0 \times 10^{-4}\) | 182 |
| \(1.0 \times 10^{-5}\) | 182 |
| \(1.0 \times 10^{-6}\) | 177 |
¿Cuál es la concentración máxima aceptable de benzoato si planea usar este electrodo selectivo de iones para analizar una muestra que contiene tan poco como salicilato de 10 —5 M con una precisión superior al 1%?
6. Watanabe y compañeros de trabajo describieron un nuevo electrodo de membrana para la determinación de cocaína, un alcaloide de base débil con una p K a de 8.64 [Watanabe, K.; Okada, K.; Oda, H.; Furuno, K.; Gomita, Y.; Katsu, T. Anal. Chim. Acta 1995, 316, 371—375]. La respuesta del electrodo para una concentración fija de cocaína es independiente del pH en el rango de 1—8, pero disminuye bruscamente por encima de un pH de 8. Ofrezca una explicación para esta dependencia del pH.
7. La Figura 11.2.14 muestra un diagrama esquemático de un electrodo enzimático que responde a la urea mediante el uso de un electrodo NH 3 sensor de gas para medir la cantidad de amoníaco liberado después de la reacción de la enzima con urea. A su vez, el electrodo NH 3 utiliza un electrodo de pH para monitorear el cambio en el pH debido al amoníaco. La respuesta del electrodo de urea viene dada por la ecuación 11.2.12. Comenzando con la ecuación 11.2.19, que da el potencial de un electrodo de pH, muestran que la ecuación 11.2.12 para el electrodo de urea es correcta.
8. Explicar por qué la respuesta de un electrodo de urea basado en NH 3 (Figura 11.2.14 y ecuación 11.2.12) es diferente de la respuesta de un electrodo de urea en el que la enzima está recubierta sobre la membrana de vidrio de un electrodo de pH (Figura 11.2.15 y ecuación 11.2 .13).
9. Un electrodo potenciométrico para HCN utiliza una membrana permeable a los gases, una solución interna tamponada de 0.01 M KAg (CN) 2 y un electrodo Ag 2 S ISE que se sumerge en la solución interna. Considerar las reacciones de equilibrio que tienen lugar dentro de la solución interna y derivar una ecuación que relaciona el potencial del electrodo con la concentración de HCN en la muestra. Para consultar tu trabajo, busca on-line la Patente US 3859191 y consulta la Figura 2.
10. Mifflin y asociados describieron un electrodo de membrana para el análisis cuantitativo de penicilina en el que la enzima penicilinasa se inmoviliza en un gel de poliacrilamida recubierto sobre la membrana de vidrio de un electrodo de pH [Mifflin, T. E.; Andriano, K. M.; Robbins, W. B. J. Chem. Educ. 1984, 61, 638—639]. Los siguientes datos se recolectaron utilizando un conjunto de estándares de penicilina.
| [penicilina] (M) | potencial (mV) |
|---|---|
| \(1.0 \times 10^{-2}\) | 220 |
| \(2.0 \times 10^{-3}\) | 204 |
| \(1.0 \times 10^{-3}\) | 190 |
| \(2.0 \times 10^{-4}\) | 153 |
| \(1.0 \times 10^{-4}\) | 135 |
| \(1.0 \times 10^{-5}\) | 96 |
| \(1.0 \times 10^{-6}\) | 80 |
a) ¿En qué rango de concentraciones hay una respuesta lineal?
b) ¿Cuál es la ecuación de la curva de calibración para este rango de concentración?
c) ¿Cuál es la concentración de penicilina en una muestra que arroja un potencial de 142 mV?
11. Un electrodo selectivo de iones se puede colocar en una celda de flujo en la que inyectamos muestras o estándares. A medida que el analito pasa a través de la célula, se registra un pico potencial en lugar de un potencial en estado estacionario. La concentración de K + en suero se ha determinado de esta manera usando estándares preparados en una matriz de NaCl 0.014 M [Meyerhoff, M. E.; Kovach, P. M. J. Chem. Educ. 1983, 9, 766—768].
| [K +] (mM) | E (unidades arb.) | [K +] (mM) | E (unidades arb.) |
|---|---|---|---|
| 0.10 | 25.5 | 0.60 | 58.7 |
| 0.20 | 37.2 | 0.80 | 64.0 |
| 0.40 | 50.8 | 1.00 | 66.8 |
Una muestra de suero de 1.00-mL se diluye a volumen en un matraz aforado de 10 mL y se analiza, dando un potencial de 51.1 (unidades arbitrarias). Reportar la concentración de K + en la muestra de suero.
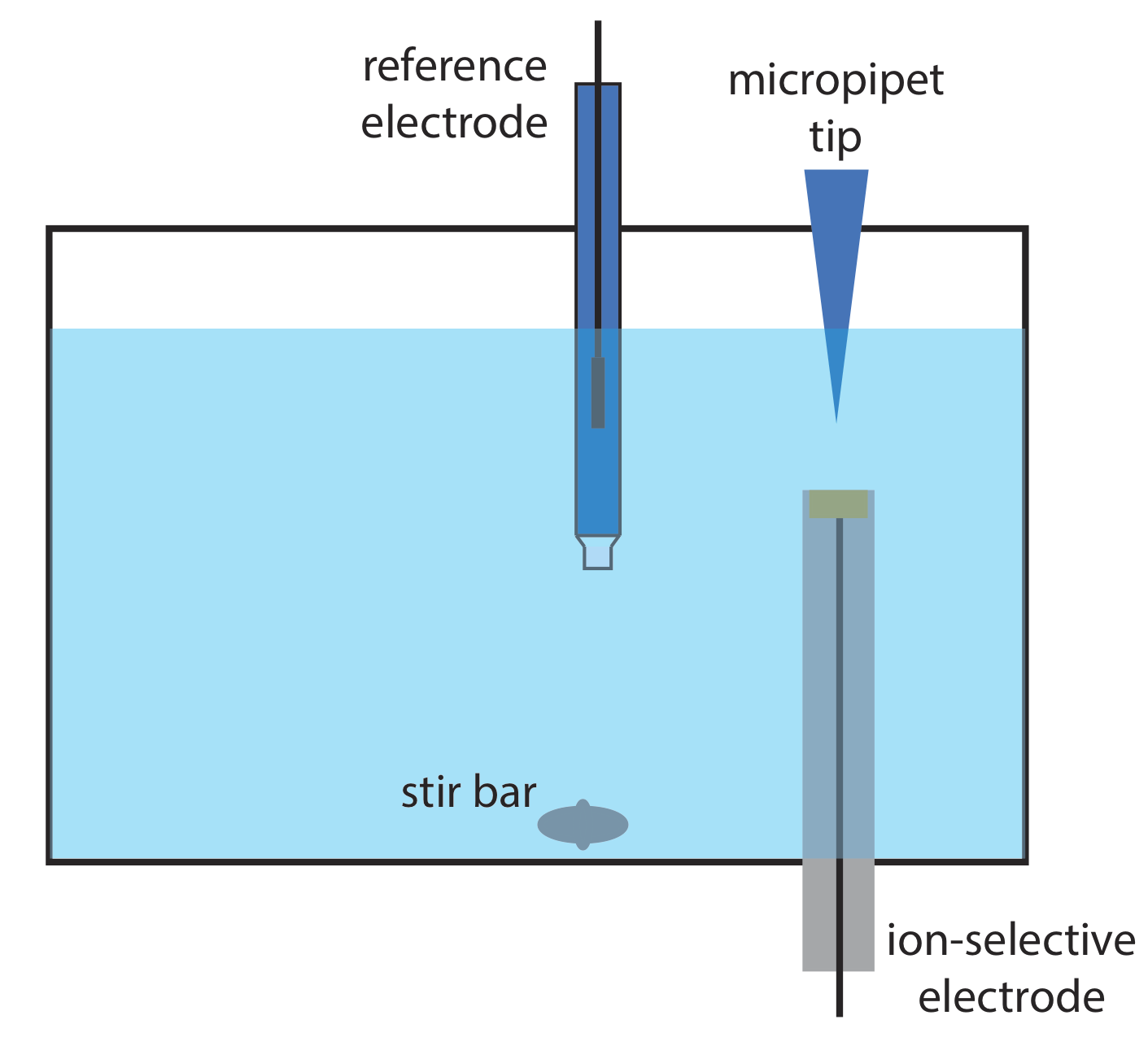
12. Wang y Taha describieron una interesante aplicación de la potenciometría, a la que llaman inyección por lotes [Wang, J.; Taha, Z. Anal. Chim. Acta 1991, 252, 215—221]. Como se muestra en la siguiente figura, un electrodo selectivo de iones se coloca en una posición invertida en un tanque de gran volumen, y se inyecta un volumen fijo de una muestra o una solución estándar hacia la superficie del electrodo usando una micropipeta. La respuesta del electrodo es un pico en el potencial que es proporcional a la concentración del analito. Los siguientes datos se recolectaron usando un electrodo de pH y un conjunto de patrones de pH.
| pH | potencial (mV) |
|---|---|
| 2.0 | +300 |
| 3.0 | +240 |
| 4.0 | +168 |
| 5.0 | +81 |
| 6.0 | +35 |
| 8.0 | —92 |
| 9.0 | —168 |
| 10.0 | —235 |
| 11.0 | —279 |
Determinar el pH de las siguientes muestras dadas los potenciales pico registrados: jugo de tomate, 167 mV; agua del grifo, —27 mV; café, 122 mV.
13. La concentración\(\text{NO}_3^-\) en una muestra de agua se determina mediante una adición estándar de un punto usando un electrodo\(\text{NO}_3^-\) selectivo de iones. Se coloca una muestra de 25.00 mL en un vaso de precipitados y se mide un potencial de 0.102 V. Se agrega una alícuota de 1.00-mL de una solución estándar de\(\text{NO}_3^-\) 200.0-mg/L, después de lo cual el potencial es 0.089 V. Reportan los mg\(\text{NO}_3^-\) /L en la muestra de agua.
14. En 1977, cuando era estudiante de pregrado en Knox College, mi compañero de laboratorio y yo completamos un experimento para determinar la concentración de fluoruro en el agua del grifo y la cantidad de flúor en la pasta de dientes. Los datos en este problema son de mi cuaderno de laboratorio.
(a) Para analizar el agua del grifo, se tomaron tres muestras de 25.0 mL y se agregaron 25.0 mL de TISAB a cada una. Medimos el potencial de cada solución usando un electrodo de referencia F — ISE y un electrodo de referencia SCE. A continuación, realizamos cinco adiciones de 1.00-mL de una solución estándar de 100.0 ppm F — a cada muestra, y midimos el potencial después de cada adición, registrando el potencial tres veces.
| mL de estándar agregado | potencial (mV), replicar 1 | potencial (mV), replicar 2 | potencial (mV), replicar 3 |
|---|---|---|---|
| 0.00 | —79 | —82 | —83 |
| 1.00 | —119 | —119 | —118 |
| 2.00 | —133 | —133 | —133 |
| 3.00 | —142 | —142 | —142 |
| 4.00 | —149 | —148 | —148 |
| 5.00 | —154 | —153 | —153 |
Reportan las partes por millón de F — en el agua del grifo.
(b) Para analizar la pasta de dientes, se midieron 0.3619 g en un matraz aforado de 100 mL, se agregaron 50.0 mL de TISAB y se diluyeron a volumen con agua destilada. Después de asegurar que la muestra estaba completamente mezclada, transferimos tres porciones de 20.0-mL a vasos de precipitados separados y midimos el potencial de cada uno usando un electrodo de referencia F — ISE y un electrodo de referencia SCE. A continuación, realizamos cinco adiciones de 1.00-mL de una solución estándar de 100.0 ppm F — a cada muestra, y midimos el potencial después de cada adición, registrando el potencial tres veces.
| mL de estándar agregado | potencial (mV), replicar 1 | potencial (mV), replicar 2 | potencial (mV), replicar 3 |
| 0.00 | —55 | —54 | —55 |
| 1.00 | —82 | —82 | —83 |
| 2.00 | —94 | —94 | —94 |
| 3.00 | —102 | —103 | —102 |
| 4.00 | —108 | —108 | —109 |
| 5.00 | —112 | —112 | —113 |
Reportan las partes por millón F — en la pasta de dientes.
15. Usted es responsable de determinar la cantidad de KI en la sal yodada y decide usar un electrodo I — selectivo de iones. Describa cómo realizaría este análisis usando estándares externos y cómo formaría este análisis usando el método de adiciones estándar.
16. Explique por qué cada uno de los siguientes factores disminuye el tiempo de análisis en la coulometría de potencial controlado: una mayor superficie para el electrodo de trabajo; un menor volumen de solución; y una velocidad de agitación más rápida.
17. La pureza de una muestra de ácido pícrico, C 6 H 3 N 3 O 7, se determina por coulometría de potencial controlado, convirtiendo el ácido pícrico en triaminofenol, C 6 H 9 N 3 O.
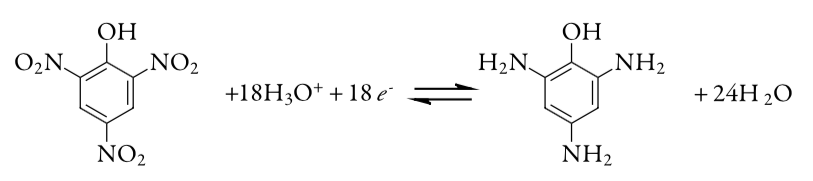
Una muestra de 0.2917 g de ácido pícrico se coloca en un matraz aforado de 1000 mL y se diluye a volumen. Una porción de 10.00 ml de esta solución se transfiere a una celda coulométrica y se agrega suficiente agua para que se sumerja el cátodo de Pt. Una electrólisis exhaustiva de la muestra re-requiere 21.67 C de carga. Reportar la pureza del ácido pícrico.
18. La concentración de H 2 S en el drenaje de una mina abandonada se determina mediante una valoración coulométrica utilizando KI como mediador y\(\text{I}_3^-\) como valorante.
\[\text{H}_{2}\text{S}(a q)+\ \mathrm{I}_{3}^{-}(a q)+2 \mathrm{H}_{2} \mathrm{O}(l)\rightleftharpoons2 \mathrm{H}_{3} \mathrm{O}^{+}(a q)+3 \mathrm{I}^{-}(a q)+\mathrm{S}(s) \nonumber\]
Se coloca una muestra de 50.00-mL de agua en una celda coulométrica, junto con un exceso de KI y una pequeña cantidad de almidón como indicador. La electrólisis se realiza a una corriente constante de 84.6 mA, requiriendo 386 s para alcanzar el punto final del almidón. Reportar la concentración de H 2 S en la muestra en μg/mL.
19. Un método para la determinación de una masa dada de H 3 AsO 3 es una valoración coulométrica\(\text{I}_3^-\) que se usa como valorante. Las reacciones y potenciales relevantes del estado estándar se resumen aquí
\[\begin{aligned} \mathrm{H}_{3} \mathrm{AsO}_{4}(a q)+2 \mathrm{H}^{+}(a q)+2 \mathrm{e}^{-} &\rightleftharpoons \ \mathrm{H}_{3} \mathrm{AsO}_{3}(a q)+\ \mathrm{H}_{2} \mathrm{O}(l) \\ \mathrm{I}_{3}^{-}(a q)+2 \mathrm{e}^{-} &\rightleftharpoons 3 \mathrm{I}^{-}(a q) \end{aligned} \nonumber\]
con potenciales de reducción de estado estándar de, respectivamente, +0.559 V y +0.536 V. Explique por qué la titulación coulométrica se realiza en una solución neutra (pH ≈ 7) en lugar de en una solución fuertemente ácida (pH < 0).
20. La producción de adiponitrilo, NC (CH 2) 4 CN, a partir de acrilonitrilo, CH 2 =CHCN, es un proceso industrial importante. Una muestra de 0.594 g de acrilonitrilo se coloca en un matraz aforado de 1 L y se diluye a volumen. Una electrólisis exhaustiva de potencial controlado de una porción de 1.00-mL del acrilonitrilo diluido requiere 1.080 C de carga. ¿Cuál es el valor de n para la reducción de acrilonitrilo a adiponitrilo?
21. El voltamograma hidrodinámico de barrido de potencial lineal para una mezcla de Fe 2 + y Fe 3 + se muestra en la siguiente figura donde i l, a e i l, c son los limitantes anódicos y catódicos corrientes.
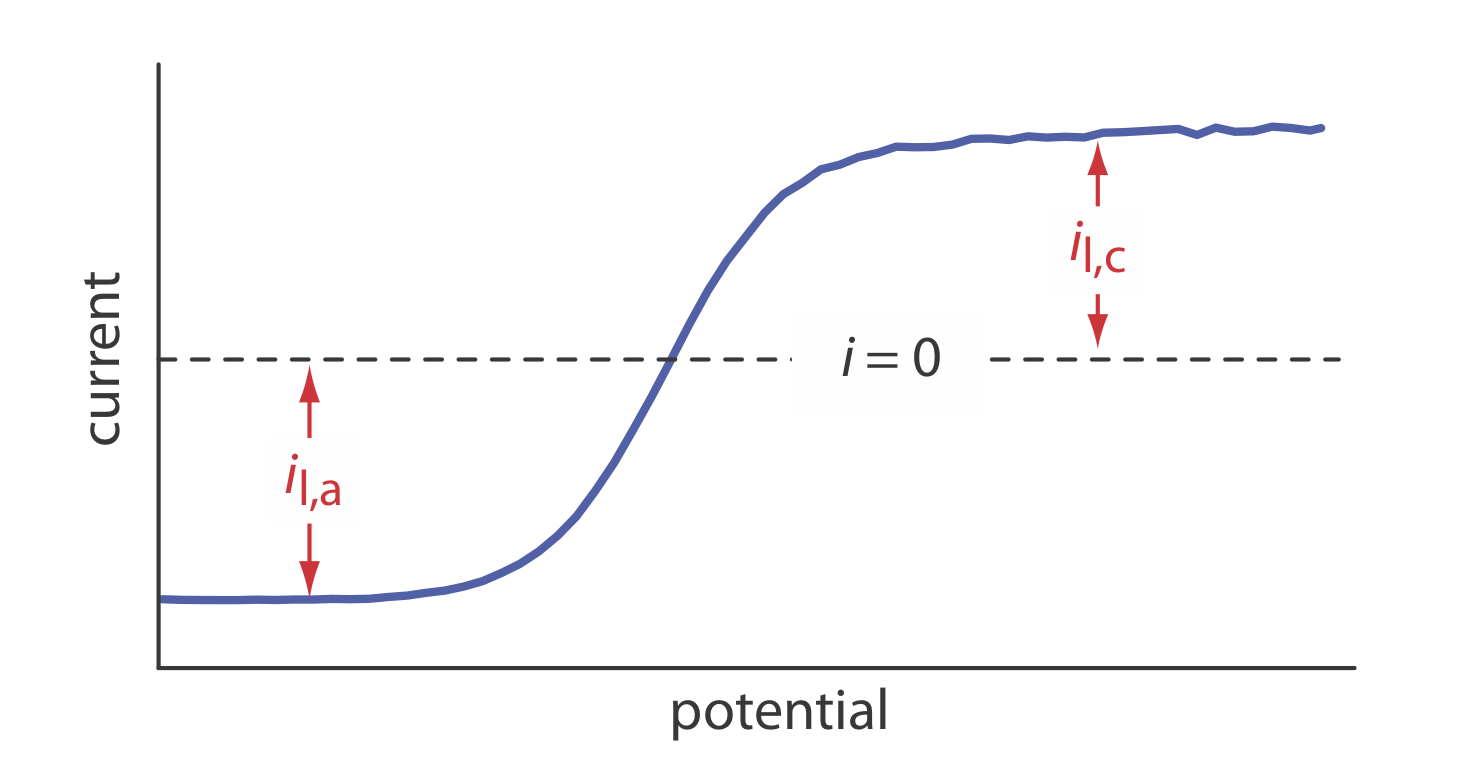
(a) Demostrar que el potencial viene dado por
\[E = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} - 0.05916 \log \frac {K_{\text{Fe}^{3+}}} {K_{\text{Fe}^{2+}}} - 0.05916 \log \frac {i - i_{l,a}}{i_{l,c} - i} \nonumber\]
(b) ¿Cuál es el potencial cuando i = 0 para una solución que es 0.100 mM Fe 3 + y 0.050 mM Fe 2 +?
22. La cantidad de azufre en los monómeros aromáticos se determina mediante polarografía diferencial de pulsos. Las soluciones estándar se preparan para su análisis disolviendo 1.000 mL del monómero purificado en 25.00 mL de un solvente electrolítico, agregando una cantidad conocida de azufre, desaireando y midiendo la corriente pico. Se obtuvieron los siguientes resultados para un conjunto de estándares de calibración.
| µg S añadido | corriente pico (µA) |
|---|---|
| 0 | 0.14 |
| 28 | 0.70 |
| 56 | 1.23 |
| 112 | 2.41 |
| 168 | 3.42 |
El análisis de una muestra de 1.000-mL, tratada de la misma manera que los estándares, da una corriente pico de 1.77 μA. Reporte los mg S/mL en la muestra.
23. La pureza de una muestra de K 3 Fe (CN) 6 se determina mediante voltamperometría hidrodinámica de barrido de potencial lineal en un electrodo de carbono vítreo. Se obtuvieron los siguientes datos para un conjunto de estándares de calibración externos.
| [K 3 Fe (CN) 6] (mM) | corriente limitadora (µA) |
|---|---|
| 2.0 | 127 |
| 4.0 | 252 |
| 6.0 | 376 |
| 8.0 | 500 |
| 10.0 | 624 |
Se prepara una muestra de K 3 Fe (CN) 6 impura para su análisis diluyendo una porción de 0.246 g a volumen en un matraz aforado de 100 mL. La corriente límite para la muestra es 444 μA. Reportar la pureza de esta muestra de K 3 Fe (CN) 6.
24. Un método para determinar si un individuo disparó recientemente una pistola es buscar rastros de antimonio en los residuos recolectados de las manos del individuo. La voltametría de extracción anódica en un electrodo de película de mercurio es ideal para este análisis. En un análisis típico se recolecta una muestra de un sospechoso utilizando un hisopo con punta de algodón humedecido con HNO 3 al 5% v/v. Después de regresar al laboratorio, el hisopo se coloca en un vial que contiene 5.0 mL de HCl 4 M que es 0.02 M en sulfato de hidrazina. Después de remojar el hisopo, una porción de 4.0-mL de la solución se transfiere a una celda electroquímica junto con 100 μL de 0.01 M HgCl 2. Después de depositar la película delgada de mercurio y antimonio, la etapa de decapado da una corriente pico de 0.38 μA. Después de agregar una adición estándar de 100 μL de\(5.00 \times 10^2\) ppb Sb, la corriente pico aumenta a 1.14 μA. ¿Cuántos nanogramos de Sb se recolectaron de la mano del sospechoso?
25. El zinc se utiliza como patrón interno en un análisis de talio mediante polarografía diferencial de pulso. Una solución estándar de\(5.00 \times 10^{-5}\) M Zn 2 + y\(2.50 \times 10^{-5}\) M Tl+ tiene corrientes pico de 5.71 μA y 3.19 μA, respectivamente. Una muestra de 8.713-g de una aleación libre de zinc se disuelve en ácido, se transfiere a un matraz aforado de 500 ml y se diluye a volumen. Una porción de 25.0 mL de esta solución se mezcla con 25.0 mL de\(5.00 \times 10^{-4}\) M Zn 2 +. El análisis de esta solución da corrientes pico de 12.3 μA y de 20.2 μA para Zn 2 + y Tl +, respectivamente. Reporta el %w/w Tl en la aleación.
26. La voltametría diferencial de pulso en un electrodo de trabajo de carbono se utiliza para determinar las concentraciones de ácido ascórbico y cafeína en formulaciones de fármacos [Lau, O.; Luk, S.; Cheung, Y. Analyst 1989, 114, 1047—1051]. En un análisis típico se tritura una tableta de 0.9183 g y se muele hasta obtener un polvo fino. Una muestra de 0.5630 g de este polvo se transfiere a un matraz aforado de 100 ml, se pone en solución y se diluye a volumen. Una porción de 0.500 mL de esta solución se transfiere luego a una celda voltamétrica que contiene 20.00 mL de un electrolito de soporte adecuado. El voltamograma resultante da corrientes pico de 1.40 μA y 3.88 μA para el ácido ascórbico y para la cafeína, respectivamente. Luego se agrega una alícuota de 0.500 ml de una solución estándar que contiene 250.0 ppm de ácido ascórbico y 200.0 ppm de cafeína. Un voltamograma de esta solución da corrientes pico de 2.80 μA y 8.02 μA para ácido ascórbico y cafeína, respectivamente. Reporte los miligramos de ácido ascórbico y miligramos de cafeína en la tableta.
27. Ratana-ohpas y compañeros de trabajo describieron un método de análisis de decapado para determinar estaño en jugos de frutas enlatados [Ratana-ohpas, R.; Kanatharana, P.; Ratana-ohpas, W.; Kongsawasdi, W. Anal. Chim. Acta 1996, 333, 115—118]. Se analizaron estándares de 50.0 ppb Sn 4 +, 100.0 ppb Sn 4 + y 150.0 ppb Sn 4 +, dando corrientes pico (unidades arbitrarias) de 83.0, 171.6 y 260.2, respectivamente. Se mezcla una muestra de 2.00-mL de jugo de lichi con 20.00 mL de 1:1 HCl/HnO 3. Se agrega una porción de 0.500-mL de esta mezcla a 10 mL de HCl 6 M y el volumen se ajusta a 30.00 mL. El análisis de esta muestra diluida dio una señal de 128.2 (unidades arbitrarias). Reportan las partes por millón Sn 4 + en la muestra original de jugo de lichi.
28. Sittampalam y Wilson describieron la preparación y uso de un sensor amperométrico para glucosa [Sittampalam, G.; Wilson, G. S. J. Chem. Educ. 1982, 59, 70—73]. El sensor se calibra midiendo la corriente de estado estacionario cuando se sumerge en soluciones estándar de glucosa. Aquí se muestra un conjunto típico de datos de calibración.
| [glucosa] (mg/100 mL) | corriente (arb. unidades) |
|---|---|
| 2.0 | 17.2 |
| 4.0 | 32.9 |
| 6.0 | 52.1 |
| 8.0 | 68.0 |
| 10.0 | 85.8 |
Una muestra de 2.00-mL se diluye a 10 mL en un matraz aforado y se mide una corriente de estado estacionario de 23.6 (unidades arbitrarias). ¿Cuál es la concentración de glucosa en la muestra en mg/100 mL?
29. La polarografía diferencial de pulso se utiliza para determinar las concentraciones de plomo, talio e indio en una mezcla. Debido a que los picos para plomo y talio, y para talio e indio se superponen, es necesario un análisis simultáneo. Las corrientes pico (en unidades arbitrarias) a —0.385 V, —0.455 V y —0.557 V se miden para una única solución estándar, y para una muestra, dando los resultados que se muestran en la siguiente tabla. Reportar los mg/mL de Pb 2 +, Tl + y In 3 + en la muestra.
| analito | [estándar] (µg/mL) | pico de corriente a —0.385 V | pico de corriente a —0.455 V | pico de corriente a —0.557 V |
|---|---|---|---|---|
| Pb 2 + | 1.0 | 26.1 | 2.9 | 0 |
| Tl + | 2.0 | 7.8 | 23.5 | 3.2 |
| En 3+ | 0.4 | 0 | 0 | 22.9 |
| muestra | 60.6 | 28.8 | 54.1 | |
30. Abass y colaboradores desarrollaron un biosensor amperométrico para\(\text{NH}_4^+\) que utiliza la enzima glutamato deshidrogenasa para catalizar la siguiente reacción
\[2 \text { - oxyglutarate }(a q)+ \ \mathrm{NH}_{4}^{+}(a q)+\mathrm{NADH}(a q)\rightleftharpoons\text { glutamate }(a q)+\ \mathrm{NAD}^{+}(a q)+\ \mathrm{H}_{2} \mathrm{O}(l) \nonumber\]
donde NADH es la forma reducida del dinucleótido de nicotinamida adenina [Abass, A. K.; Hart, J. P.; Cowell, D. C.; Chapell, A. Anal. Chim. Acta 1988, 373, 1—8]. El biosensor realmente responde a la concentración de NADH, sin embargo, la velocidad de la reacción depende de la concentración de\(\text{NH}_4^+\). Si las concentraciones iniciales de 2-oxiglutarato y NADH son las mismas para todas las muestras y estándares, entonces la señal es proporcional a la concentración de\(\text{NH}_4^+\). Como se muestra en la siguiente tabla, la sensibilidad del método depende del pH.
| pH | sensibilidad (nA S —1 M —1) |
|---|---|
| 6.2 | \(1.67 \times 10^3\) |
| 6.75 | \(5.00 \times 10^3\) |
| 7.3 | \(9.33 \times 10^3\) |
| 7.7 | \(1.04 \times 10^4\) |
| 8.3 | \(1.27 \times 10^4\) |
| 9.3 | \(2.67 \times 10^3\) |
Dos posibles explicaciones para el efecto del pH en la sensibilidad de este análisis son la química ácido-base\(\text{NH}_4^+\) y la química ácido-base de la enzima. Dado que la p K a para\(\text{NH}_4^+\) es 9.244, explique la fuente de esta sensibilidad dependiente del pH.
31. El esquema de especiación para metales traza en el Cuadro 11.4.2 los divide en siete grupos definidos operacionalmente mediante la recolección y análisis de dos muestras después de cada uno de los cuatro tratamientos, requiriendo un total de ocho muestras y ocho mediciones. Después de eliminar las partículas insolubles por filtración (tratamiento 1), la solución se analiza para determinar la concentración de metales lábiles ASV y para la concentración total de metales. Una porción de la solución filtrada se pasa a través de una columna de intercambio iónico (tratamiento 2), y se determinan las concentraciones de metal ASV y de metal total. Una segunda porción de la solución filtrada se irradia con luz UV (tratamiento 3), y se miden las concentraciones de metal ASV y de metal total. Finalmente, una tercera porción de la solución filtrada se irradia con luz UV y se pasa a través de una columna de intercambio iónico (tratamiento 4), y se determinan las concentraciones de metal lábil ASV y de metal total nuevamente. Los grupos que se incluyen en cada medición se resumen en la siguiente tabla.
| tratamiento | grupos eliminados por tratamiento | grupos que contribuyen a metales lábiles a ASV | grupos que contribuyen al total de metales |
|---|---|---|---|
| 1 | ninguno | I, II, III | I, II, III, IV, V, VI, VII |
| 2 | I, IV, V | II, III | II, III, V1, VII |
| 3 | ninguno | I, II, III, IV, VI | I, II, III, IV, V, VI, VII |
| 4 | I, II, IV, V, VI | III | III, VII |
(a) Explique cómo puede utilizar estas ocho mediciones para determinar la concentración de metales presentes en cada uno de los siete grupos identificados en el Cuadro 11.4.2.
b) Batley y Florence reportan los siguientes resultados para la especiación de cadmio, plomo y cobre en una muestra de agua de mar [Batley, G. E.; Florence, T. M. Anal. A lett. 1976, 9, 379—388]. Determine la especiación de cada metal en comentario sobre sus resultados.
|
medición tratamiento: ASV-lábil o total |
ppb Cd 2+ | ppb Pb 2+ | ppb Cu 2+ |
|---|---|---|---|
| 1: ASV-lábil | 0.24 | 0.39 | 0.26 |
| 2: total | 0.28 | 0.50 | 0.40 |
| 2: ASV-lábil | 0.21 | 0.33 | 0.17 |
| 2: total | 0.26 | 0.43 | 0.24 |
| 3: ASV-lábil | 0.26 | 0.37 | 0.33 |
| 3: total | 0.28 | 0.5 | 0.43 |
| 4: ASV-lábil | 0.00 | 0.00 | 0.00 |
| 4: total | 0.02 | 0.12 | 0.10 |
32. La concentración de Cu 2 + en el agua de mar se determina mediante voltametría de extracción anódica en un electrodo de gota de mercurio colgante después de liberar primero cualquier cobre unido a la materia orgánica. A una muestra de 20.00 mL de agua de mar se le agrega 1 mL de 0.05 M HNO 3 y 1 mL de 0.1% H 2 O 2. La muestra se irradia con luz UV durante 8 h y luego se diluye a volumen en un matraz aforado de 25 ml. La deposición de Cu 2 + se realiza a —0.3 V versus un SCE por 10 min, produciendo una corriente pico de 26.1 (unidades arbitrarias). Una segunda muestra de 20.00 mL del agua de mar se trata de manera idéntica, excepto que se agrega 0.1 mL de una solución de 5.00 μM de Cu 2 +, produciendo una corriente pico de 38.4 (unidades arbitrarias). Reportar la concentración de Cu 2 + en el agua de mar en mg/L.
33. Los fármacos de tioamida se determinan mediante análisis de extracción catódica [Davidson, I. E.; Smyth, W. F. Anal. Chem. 1977, 49, 1195—1198]. La deposición ocurre a +0.05 V versus un SCE. Durante la etapa de extracción se escanea catódicamente el potencial y se observa un pico de extracción a —0.52 V. En una aplicación típica se mezcla una muestra de orina de 2.00-mL con 2.00 mL de un tampón pH 4.78. Después de una deposición de 2.00 min, se mide una corriente pico de 0.562 μA. A la misma solución se le agrega una adición de 0.10 mL de una solución de 5.00 μM del medicamento. Se registra una corriente pico de 0.837 μA usando las mismas condiciones de deposición y decapado. Reporte la concentración molar del medicamento en la muestra de orina.
34. La concentración de vanadio (V) en el agua de mar se determina mediante voltamperometría de extracción adsortiva después de formar un complejo con catecol [van der Berg, C. M. G.; Huang, Z. Q. Anal. Chem. 1984, 56, 2383—2386]. El complejo catecol-V (V) se deposita sobre un electrodo de gota de mercurio colgante a un potencial de —0.1 V frente a un electrodo de referencia Ag/AgCl. Un barrido de potencial catódico da un pico de extracción que es proporcional a la concentración de V (V). Se utilizan las siguientes adiciones estándar para analizar una muestra de agua de mar.
| [V (V)] agregado (M) | corriente pico (µA) |
|---|---|
|
\(2.0 \times 10^{-8}\) |
24 |
| \(4.0 \times 10^{-8}\) | 33 |
| \(8.0 \times 10^{-8}\) | 52 |
| \(1.2 \times 10^{-7}\) | 69 |
| \(1.8 \times 10^{-7}\) | 97 |
| \(2.8 \times 10^{-7}\) | 140 |
Determinar la concentración molar de V (V) en la muestra de agua de mar, asumiendo que las adiciones estándar dan como resultado un cambio insignificante en el volumen de la muestra.
35. El potencial de reducción del estado estándar para Cu 2 + a Cu es +0.342 V frente a la SHE. Dado que Cu 2 + forma un complejo muy estable con el ligando EDTA, ¿espera que el potencial de reducción en estado estándar para Cu (EDTA) 2— sea mayor que +0.342 V, menor que +0.342 V, o igual a +0.342 V? Explica tu razonamiento.
36. Los potenciales polarográficos de media onda (versus el SCE) para Pb 2+ y para Tl + en HCl 1 M son, respectivamente, —0.44 V y —0.45 V. En un electrolito de NaOH 1 M, sin embargo, los potenciales de media onda son —0.76 V para Pb 2 + y —0.48 V para Tl +. ¿Por qué el cambio en el electrolito tiene un efecto tan significativo en el potencial de media onda para Pb 2 +, pero no en el potencial de media onda para Tl +?
37. Los siguientes datos para la reducción de Pb 2 + fueron recolectados por polarografía de pulso normal.
| potencial (V vs. SCE) | corriente (µA) |
|---|---|
| —0.345 | 0.16 |
| —0.370 | 0.98 |
| —0.383 | 2.05 |
| —0.393 | 3.13 |
| —0.409 | 4.62 |
| —0.420 | 5.16 |
La corriente límite fue de 5.67 μA. Verificar que la reacción de reducción sea reversible y determinar los valores para n y E 1/2. Los potenciales de media onda para los polarogramas de pulso normal de Pb 2 + en presencia de varias concentraciones diferentes de OH, se muestran en la siguiente tabla.
| [OH —] (M) | \(E_{1/2}\)(V vs. SCE) | [OH —] (M) | \(E_{1/2}\)(V vs. SCE) |
|---|---|---|---|
| 0.050 | \ (E_ {1/2}\) (V vs SCE) ">—0.646 | 0.150 | \ (E_ {1/2}\) (V vs SCE) ">—0.689 |
| 0.100 | \ (E_ {1/2}\) (V frente a SCE) ">—0.673 | 0.300 | \ (E_ {1/2}\) (V frente a SCE) ">—0.715 |
Determinar la estequiometría del complejo PB-hidróxido y su constante de formación.
38. En 1977, cuando era estudiante de pregrado en Knox College, mi compañero de laboratorio y yo completamos un experimento para estudiar el comportamiento voltamétrico de Cd 2 + (en 0.1 M KNO 3) y Ni 2 + (en 0.2 M KNO 3) en un electrodo de mercurio de caída. Los datos en este problema son de mi cuaderno de laboratorio. Todos los potenciales son relativos a un electrodo de referencia SCE.
| potencial para Cd 2+ (V) | corriente para Cd 2+ (µA) | potencial para Ni 2+ (V) | corriente para Ni 2+ (µA) |
|---|---|---|---|
| —0.60 | 4.5 | —1.07 | 1.90 |
| —0.58 | 3.4 | —1.05 | 1.75 |
| —0.56 | 2.1 | —1.03 | 1.50 |
| —0.54 | 0.6 | —1.02 | 1.25 |
| —0.52 | 0.2 | —1.00 | 1.00 |
Las corrientes limitantes para Cd 2 + fueron 4.8 μA y las de Ni 2 + fueron 2.0 μA. Evalúe la reversibilidad electroquímica para cada ion metálico y comente sus resultados.
39. Baldwin y sus colaboradores reportan los siguientes datos de un estudio de voltamperometría cíclica del comportamiento electroquímico de p-fenilendiamina en un tampón pH 7 [Baldwin, R. P.; Ravichandran, K.; Johnson, R. K. J. Chem. Educ. 1984, 61, 820—823]. Todos los potenciales se miden en relación con un SCE.
| velocidad de exploración (MV/s) | E p, a (V) | E p, c (V) | i p, a (mA) | i p, c (mA) |
| 2 | 0.148 | 0.104 | 0.34 | 0.30 |
| 5 | 0.149 | 0.098 | 0.56 | 0.53 |
| 10 | 0.152 | 0.095 | 1.00 | 0.04 |
| 20 | 0.161 | 0.095 | 1.44 | 1.44 |
| 50 | 0.167 | 0.082 | 2.12 | 1.81 |
| 100 | 0.180 | 0.063 | 2.50 | 2.19 |
El barrido inicial es hacia potenciales más positivos, lo que lleva a la reacción de oxidación que se muestra aquí.
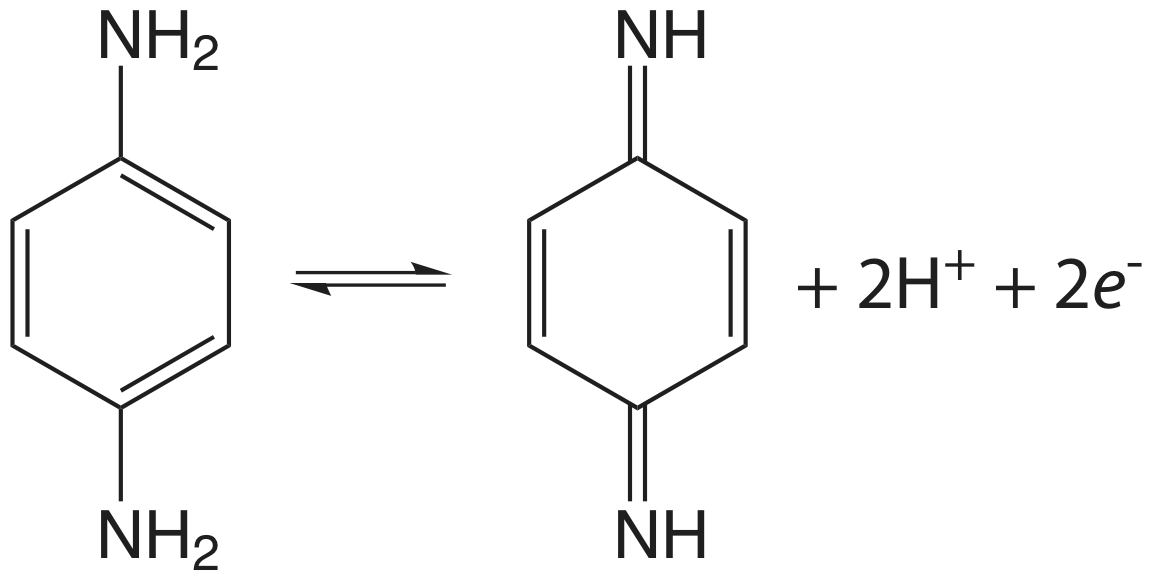
Utilice estos datos para demostrar que la reacción es electroquímicamente irreversible. Una reacción puede mostrar irreversibilidad electroquímica debido a la lenta cinética de transferencia de electrones o porque el producto de la reacción de oxidación participa en una reacción química que produce una especie no electroactiva. Con base en los datos de este problema, ¿cuál es la fuente probable de la irreversibilidad electroquímica de la p-fenilendiamina?