12.1: Visión general de las separaciones analíticas
- Page ID
- 75615

En el Capítulo 7 se examinaron varios métodos para separar un analito de potenciales interferentes. Por ejemplo, en una extracción líquido-líquido el analito y el interferente inicialmente están presentes en una sola fase líquida. Añadimos una segunda fase líquida inmiscible y las mezclamos a fondo agitando. Durante este proceso el analito y los interferentes se reparten entre las dos fases en diferentes grados, efectuando su separación. Después de dejar que las fases se separen, extraemos la fase enriquecida en analito. A pesar del poder de las extracciones líquido-líquido, existen limitaciones significativas.
Dos limitaciones de las extracciones líquido-líquido
Supongamos que tenemos una muestra que contiene un analito en una matriz que es incompatible con nuestro método analítico. Para determinar la concentración del analito primero lo separamos de la matriz mediante una simple extracción líquido-líquido. Si tenemos varios analitos, es posible que necesitemos completar una extracción separada para cada analito. Para una mezcla compleja de analitos esto rápidamente se convierte en un proceso tedioso. Esta es una limitación para una extracción líquido-líquido.
Una limitación más significativa es que la extensión de una separación depende de la proporción de distribución de cada especie en la muestra. Si la relación de distribución del analito es similar a la de otra especie, entonces su separación se vuelve imposible. Por ejemplo, supongamos que un analito, A, y un interferente, I, tienen relaciones de distribución de, respectivamente, 5 y 0.5. Si utilizamos una extracción líquido-líquido con volúmenes iguales de muestra y extractante, entonces es fácil demostrar que una sola extracción elimina aproximadamente 83% del analito y 33% del interferente. Aunque podemos eliminar el 99% del analito con tres extracciones, también eliminamos el 70% del interferente. De hecho, no existe una combinación práctica de número de extracciones o volúmenes de muestra y extractante que produzcan una separación aceptable.
Del Capítulo 7 sabemos que la relación de distribución, D, para un soluto, S, es
\[D=\frac{[S]_{\mathrm{ext}}}{[S]_{\mathrm{samp}}} \nonumber\]
donde [S] ext es su concentración de equilibrio en la fase de extracción y [S] samp es su concentración de equilibrio en la muestra. Podemos usar la relación de distribución para calcular la fracción de S que queda en la muestra, q samp, después de una extracción
\[q_{\text {samp}}=\frac{V_{\text {samp }}}{D V_{\text {ext }}+V_{\text {samp }}} \nonumber\]
donde V samp es el volumen de muestra y V ext es el volumen de la fase de extracción. Por ejemplo, si D = 10, V samp = 20 y V ext = 5, la fracción de S que queda en la muestra después de la extracción es
\[q_{\text { sanp }}=\frac{20}{10 \times 5+20}=0.29 \nonumber\]
o 29%. El 71% restante del analito se encuentra en la fase de extracción.
Una mejor manera de separar mezclas
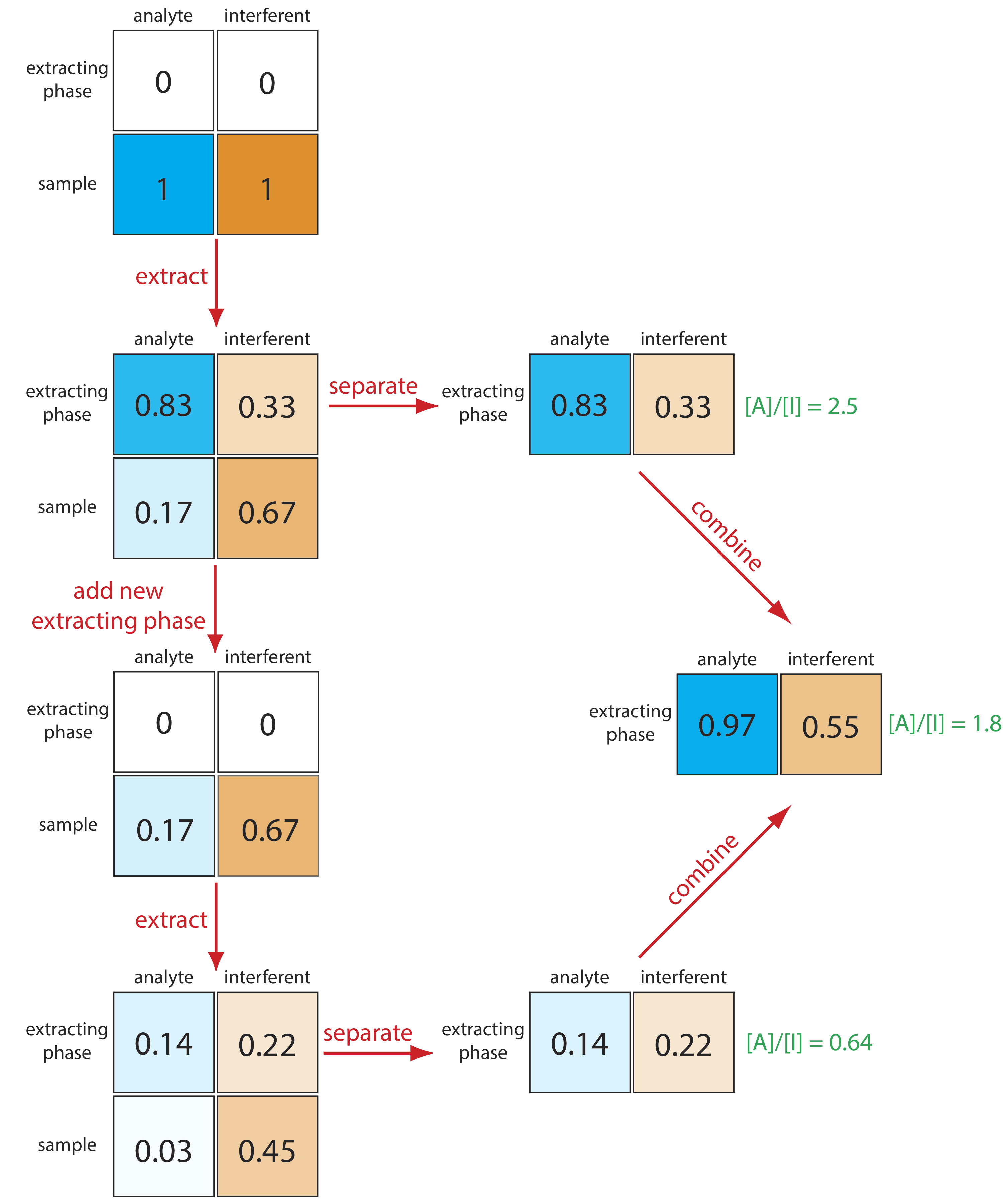
El problema con una extracción líquido-líquido es que la separación se produce en una sola dirección: de la muestra a la fase de extracción. Echemos un vistazo más de cerca a la extracción líquido-líquido de un analito y un interferente con relaciones de distribución de, respectivamente, 5 y 0.5. La Figura 12.1.1 muestra que una sola extracción utilizando volúmenes iguales de muestra y extractante transfiere 83% del analito y 33% del interferente a la fase de extracción. Si las concentraciones originales de A e I son idénticas, entonces su relación de concentración en la fase de extracción después de una extracción es
\[\frac{[A]}{[I]}=\frac{0.83}{0.33}=2.5 \nonumber\]
Una sola extracción, por lo tanto, enriquece el analito por un factor de\(2.5 \times\). Después de completar una segunda extracción (Figura 12.1.1 ) y combinar las dos fases de extracción, la separación del analito y el interferente, sorprendentemente, es menos eficiente.
\[\frac{[A]}{[I]}=\frac{0.97}{0.55}=1.8 \nonumber\]
La figura 12.1.1 deja claro por qué la segunda extracción da como resultado una separación general más pobre: ¡la segunda extracción realmente favorece al interferente!
Podemos mejorar la separación extrayendo primero los solutos de la muestra a la fase de extracción y luego extrayéndolos de nuevo en una porción fresca de disolvente que coincida con la matriz de la muestra (Figura 12.1.2 ). Debido a que el analito tiene la relación de distribución más grande, más de él se mueve hacia el extractante durante la primera extracción y menos vuelve a la fase de muestra durante la segunda extracción. En este caso la relación de concentración en la fase de extracción después de dos extracciones es significativamente mayor.
\[\frac{[A]}{[I]}=\frac{0.69}{0.11}=6.3 \nonumber\]
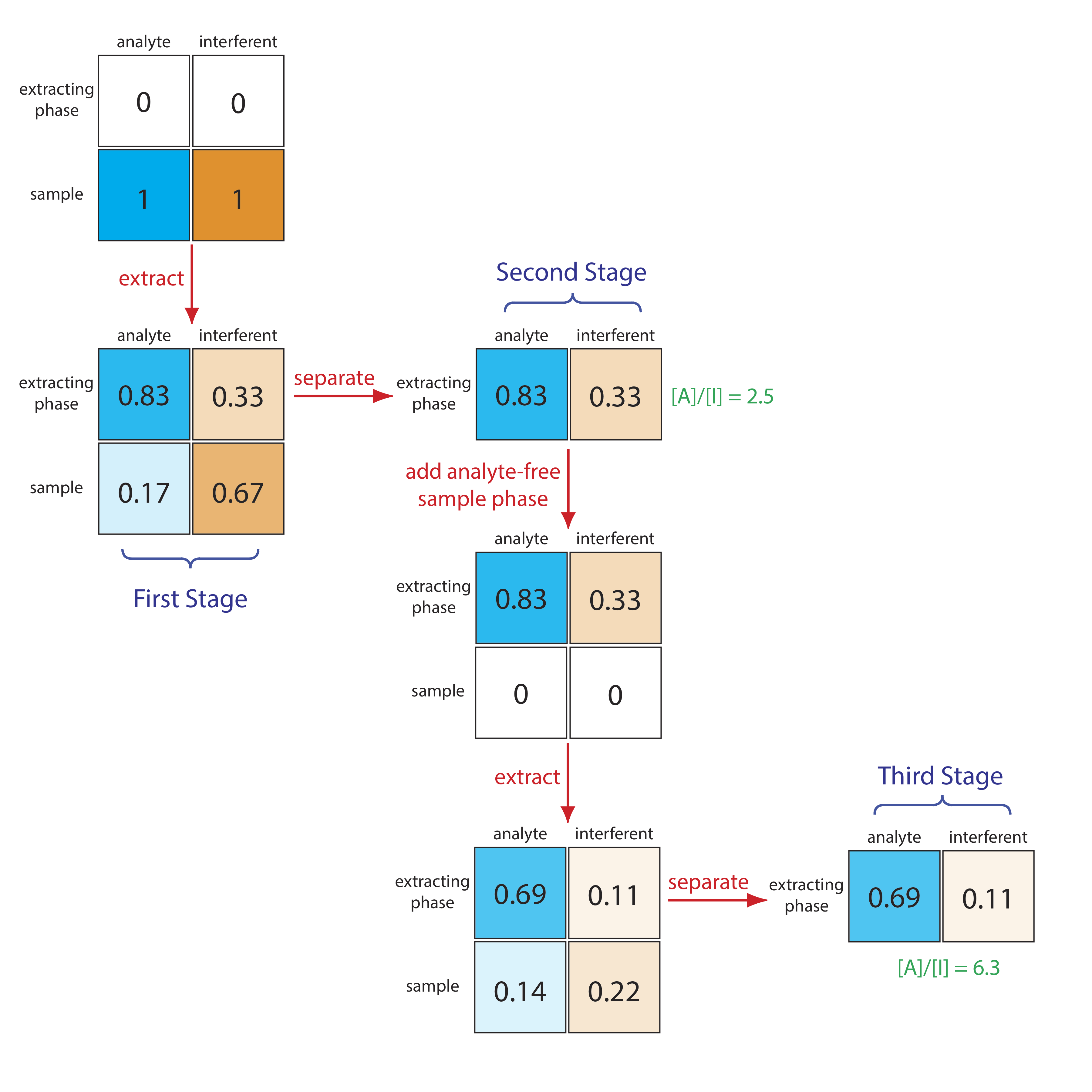
No se muestra en la Figura 12.2 es que podemos agregar una porción fresca de la fase de extracción a la muestra que queda después de la primera extracción (la fila inferior de la primera etapa en la Figura 12.2, iniciando el proceso de nuevo. A medida que aumentamos el número de extracciones, el analito y el interferente se extienden cada uno en el espacio a lo largo de una serie de etapas. Debido a que la proporción de distribución del interferente es menor que la del analito, el interferente se queda atrás del analito. Con un número suficiente de extracciones, es decir, un número suficiente de etapas, es posible la separación completa del analito y el interferente. Este proceso de extracción de los solutos de ida y vuelta entre porciones frescas de las dos fases, que llamamos extracción a contracorriente, fue desarrollado por Craig en la década de 1940 [Craig, L. C. J. Biol. Chem. 1944, 155, 519—534]. El mismo fenómeno forma la base de la cromatografía moderna.
Consulte el Apéndice 16 para una consideración más detallada de las matemáticas detrás de una extracción en contracorriente.
Separaciones Cromatográficas
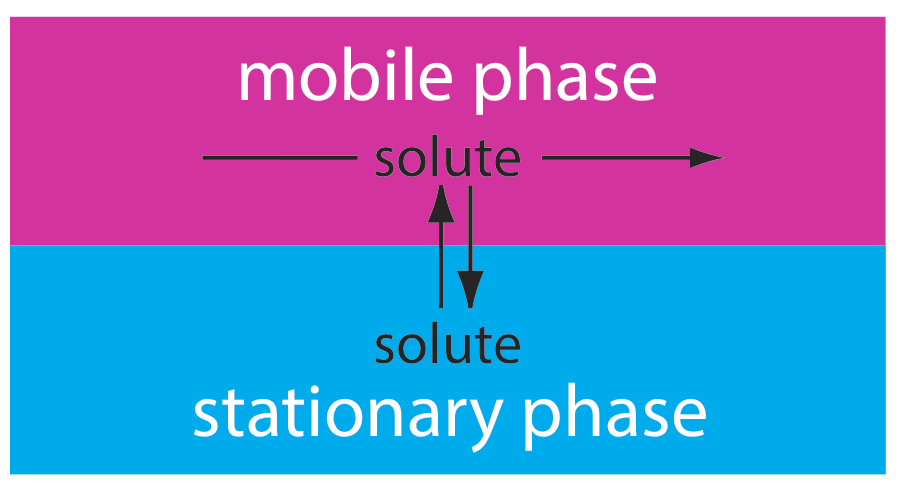
En cromatografía pasamos una fase libre de muestra, que llamamos la fase móvil, sobre una segunda fase estacionaria sin muestra que permanece fija en el espacio (Figura 12.1.3 ). Inyectamos o colocamos la muestra en la fase móvil. A medida que la muestra se mueve con la fase móvil, sus componentes se reparten entre la fase móvil y la fase estacionaria. Un componente cuya relación de distribución favorece la fase estacionaria requiere más tiempo para pasar por el sistema. Dado el tiempo suficiente y la suficiente fase estacionaria y móvil, podemos separar los solutos aunque tengan relaciones de distribución similares.
Hay muchas maneras en las que podemos identificar una separación cromatográfica: describiendo el estado físico de la fase móvil y la fase estacionaria; describiendo cómo ponemos la fase estacionaria y la fase móvil en contacto entre sí; o describiendo las interacciones químicas o físicas entre el soluto y la fase estacionaria. Consideremos brevemente cómo podríamos usar cada una de estas clasificaciones.
Podemos rastrear la historia de la cromatografía hasta el cambio de siglo cuando el botánico ruso Mikhail Tswett utilizó una columna llena de carbonato de calcio y una fase móvil de éter de petróleo para separar los pigmentos coloreados de los extractos de plantas. A medida que la muestra se movía a través de la columna, los pigmentos de la planta se separaron en bandas coloreadas individuales Después de efectuar la separación, el carbonato de calcio se retiró de la columna, se seccionó y se recuperaron los pigmentos. Tswett nombró a la técnica cromatografía, combinando las palabras griegas para “color” y “escribir”. Hubo poco interés en la técnica de Tswett hasta que Martin y Synge fueron pioneros en el desarrollo de una teoría de la cromatografía (ver Martin, A. J. P.; Synge, R. L. M. “A New Form of Chromatogram Emploating Two Liquid Phases”, Biochem. J. 1941, 35, 1358—1366). Martin y Synge fueron galardonados con el Premio Nobel de Química 1952 por esta obra.
Tipos de Fases Móviles y Fases Estacionarias
La fase móvil es un líquido o un gas, y la fase estacionaria es una película sólida o líquida recubierta sobre un sustrato sólido. A menudo nombramos técnicas cromatográficas enumerando el tipo de fase móvil seguida del tipo de fase estacionaria. En la cromatografía gas-líquido, por ejemplo, la fase móvil es un gas y la fase estacionaria es una película líquida recubierta sobre un sustrato sólido. Si el nombre de una técnica incluye solo una fase, como en la cromatografía de gases, es la fase móvil.
Contacto entre la fase móvil y la fase estacionaria
Existen dos métodos comunes para poner en contacto la fase móvil y la fase estacionaria. En cromatografía en columna empaquetamos la fase estacionaria en una columna estrecha y pasamos la fase móvil a través de la columna usando gravedad o aplicando presión. La fase estacionaria es una partícula sólida o una película líquida delgada recubierta sobre un material de relleno particulado sólido o sobre las paredes de la columna.
En la cromatografía plana, la fase estacionaria se recubre sobre una superficie plana, típicamente, una placa de vidrio, metal o plástico. Un extremo de la placa se coloca en un depósito que contiene la fase móvil, que se mueve a través de la fase estacionaria por acción capilar. En la cromatografía en papel, por ejemplo, el papel es la fase estacionaria.
Interacción entre el soluto y la fase estacionaria
La interacción entre el soluto y la fase estacionaria proporciona un tercer método para describir una separación (Figura 12.1.4 ). En la cromatografía de adsorción, los solutos se separan en función de su capacidad para adsorberse a una fase estacionaria sólida. En la cromatografía de reparto, la fase estacionaria es una película líquida delgada sobre un soporte sólido. La separación ocurre porque existe una diferencia en el reparto de equilibrio de solutos entre la fase estacionaria y la fase móvil. Una fase estacionaria que consiste en un soporte sólido con grupos funcionales aniónicos (por ejemplo\(-\text{SO}_3^-\)) o catiónicos unidos covalentemente es la base para la cromatografía de intercambio iónico en la que los solutos iónicos son atraídos a la fase estacionaria por fuerzas electrostáticas.\(-\text{N(CH}_3)_3^+\) En la cromatografía de exclusión por tamaño la fase estacionaria es una partícula porosa o gel, con separación basada en el tamaño de los solutos. Los solutos más grandes son incapaces de penetrar tan profundamente en la fase estacionaria porosa y pasar más rápidamente a través de la columna.
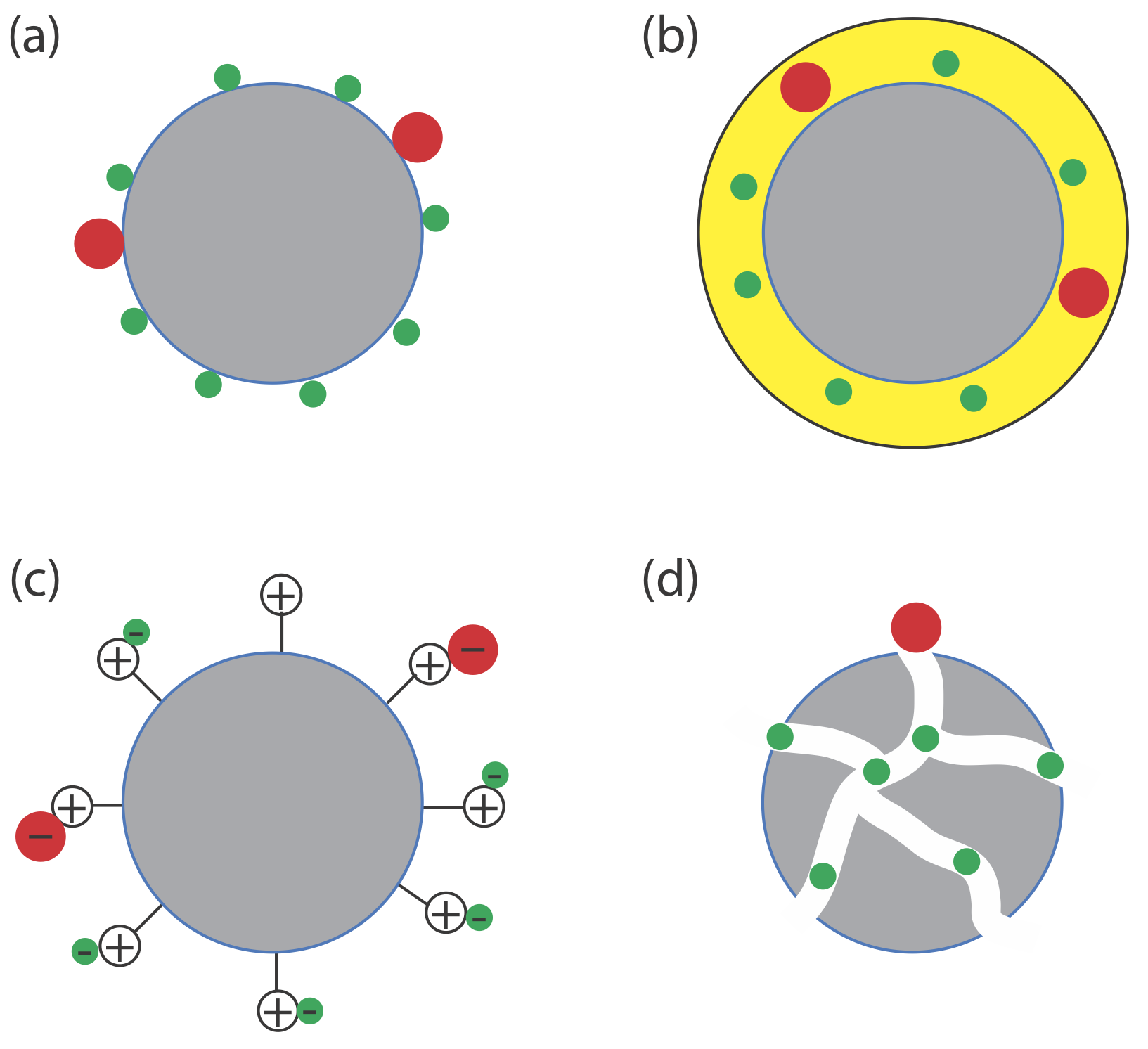
Hay otras interacciones que pueden servir como base de una separación. En la cromatografía de afinidad la interacción entre un antígeno y un anticuerpo, entre una enzima y un sustrato, o entre un receptor y un ligando forma la base de una separación. Consulte los recursos adicionales de este capítulo para algunas lecturas sugeridas.
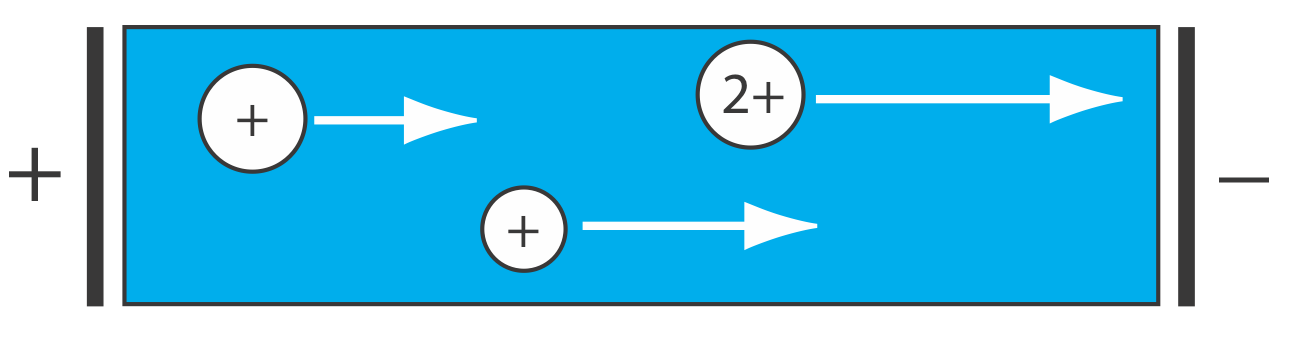
Separaciones electroforéticas
En cromatografía, se produce una separación porque existe una diferencia en el reparto de equilibrio de solutos entre la fase móvil y la fase estacionaria. La partición en equilibrio, sin embargo, no es la única base para efectuar una separación. En una separación electroforética, por ejemplo, los solutos cargados migran bajo la influencia de un potencial aplicado. Se produce una separación por diferencias en las cargas y los tamaños de los solutos (Figura 12.1.5 ).