
12.2: Teoría General de la Cromatografía en Columna
- Page ID
- 75628

De los dos métodos para poner en contacto la fase estacionaria y las fases móviles, el más importante es la cromatografía en columna. En esta sección desarrollamos una teoría general que podemos aplicar a cualquier forma de cromatografía en columna.
La Figura 12.2.1 proporciona una vista simple de un experimento de cromatografía en columna líquido-sólido. La muestra se introduce como una banda estrecha en la parte superior de la columna. Idealmente, el perfil de concentración inicial del soluto es rectangular (Figura 12.2.2 a). A medida que la muestra se mueve hacia abajo por la columna, los solutos comienzan a separarse (Figura 12.2.1 b, c) y las bandas individuales de solutos comienzan a ampliarse y desarrollar un perfil gaussiano (Figura 12.2.2 b, c). Si la fuerza de la interacción de cada soluto con la fase estacionaria es suficientemente diferente, entonces los solutos se separan en bandas individuales (Figura 12.2.1 d y Figura 12.2.2 d).
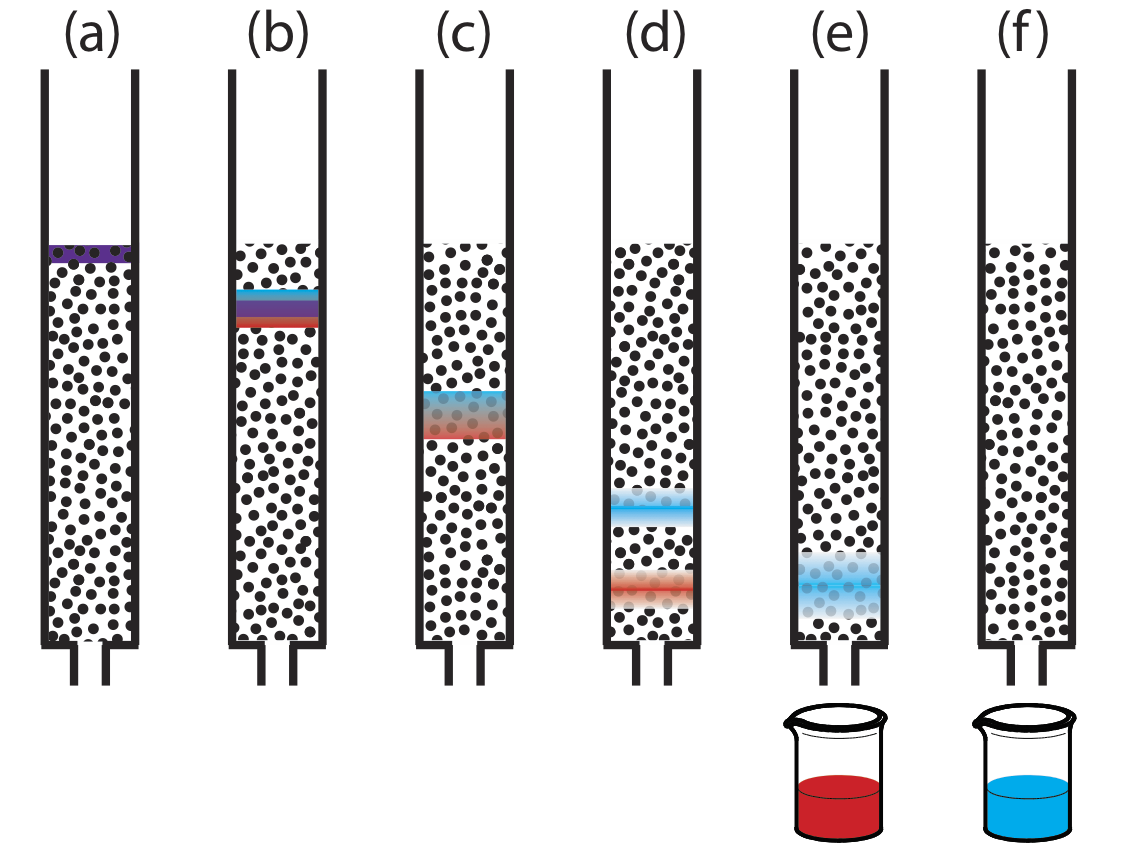
|
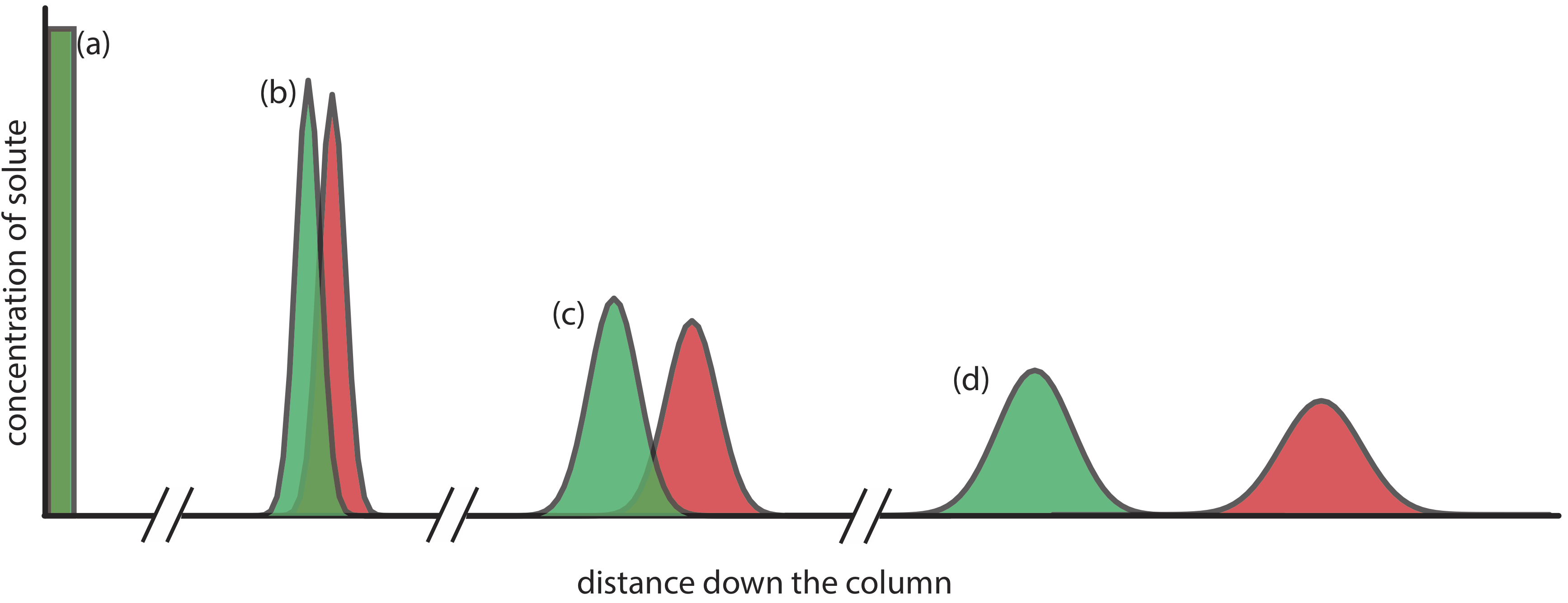
Figura 12.2.2 . Una vista alternativa de la separación en la Figura 12.2.1 que muestra la concentración de cada soluto en función de la distancia por la columna. |
Podemos seguir el progreso de la separación recogiendo fracciones a medida que eluyen de la columna (Figura 12.2.1 e, f), o colocando un detector adecuado al final de la columna. Una gráfica de la respuesta del detector en función del tiempo de elución, o en función del volumen de la fase móvil, se conoce como cromatograma (Figura 12.2.3 ), y consiste en un pico para cada soluto.
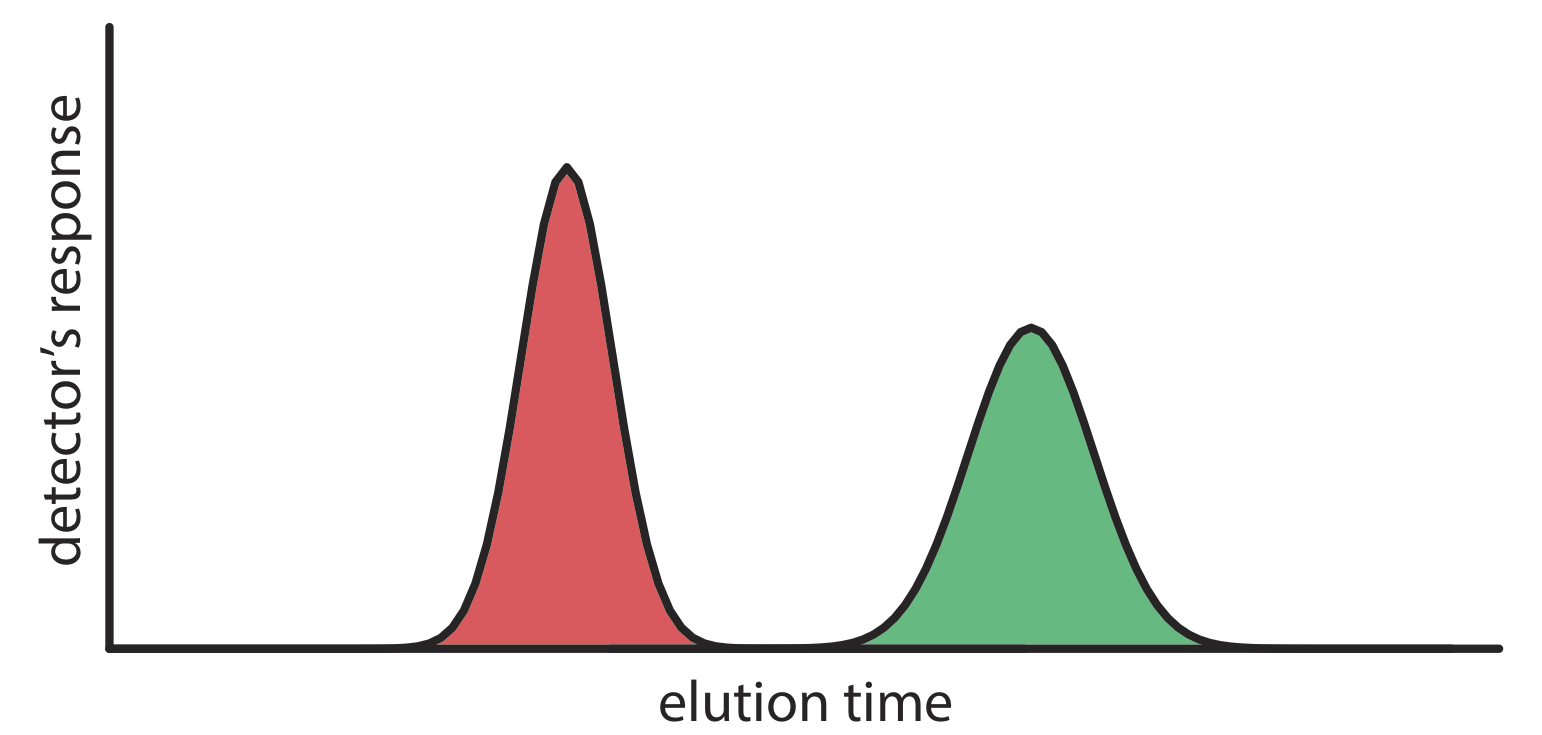
Hay muchos detectores posibles que podemos usar para monitorear la separación. Secciones posteriores de este capítulo describen algunas de las más populares.
Podemos caracterizar las propiedades de un pico cromatográfico de varias maneras, dos de las cuales se muestran en la Figura 12.2.4 . El tiempo de retención, t r, es el tiempo entre la inyección de la muestra y la respuesta máxima para el pico del soluto. El ancho basal de un pico cromatográfico, w, como se muestra en la Figura 12.2.4 , se determina extendiendo las líneas tangentes desde los puntos de inflexión a cada lado del pico hasta la línea base. Aunque usualmente reportamos t r y w usando unidades de tiempo, podemos reportarlos usando unidades de volumen multiplicando cada uno por la velocidad de la fase móvil, o reportarlos en unidades lineales midiendo distancias con una regla.
Por ejemplo, el volumen de retención de un soluto, V r, es\(t_\text{r} \times u\) donde u es la velocidad de la fase móvil a través de la columna.
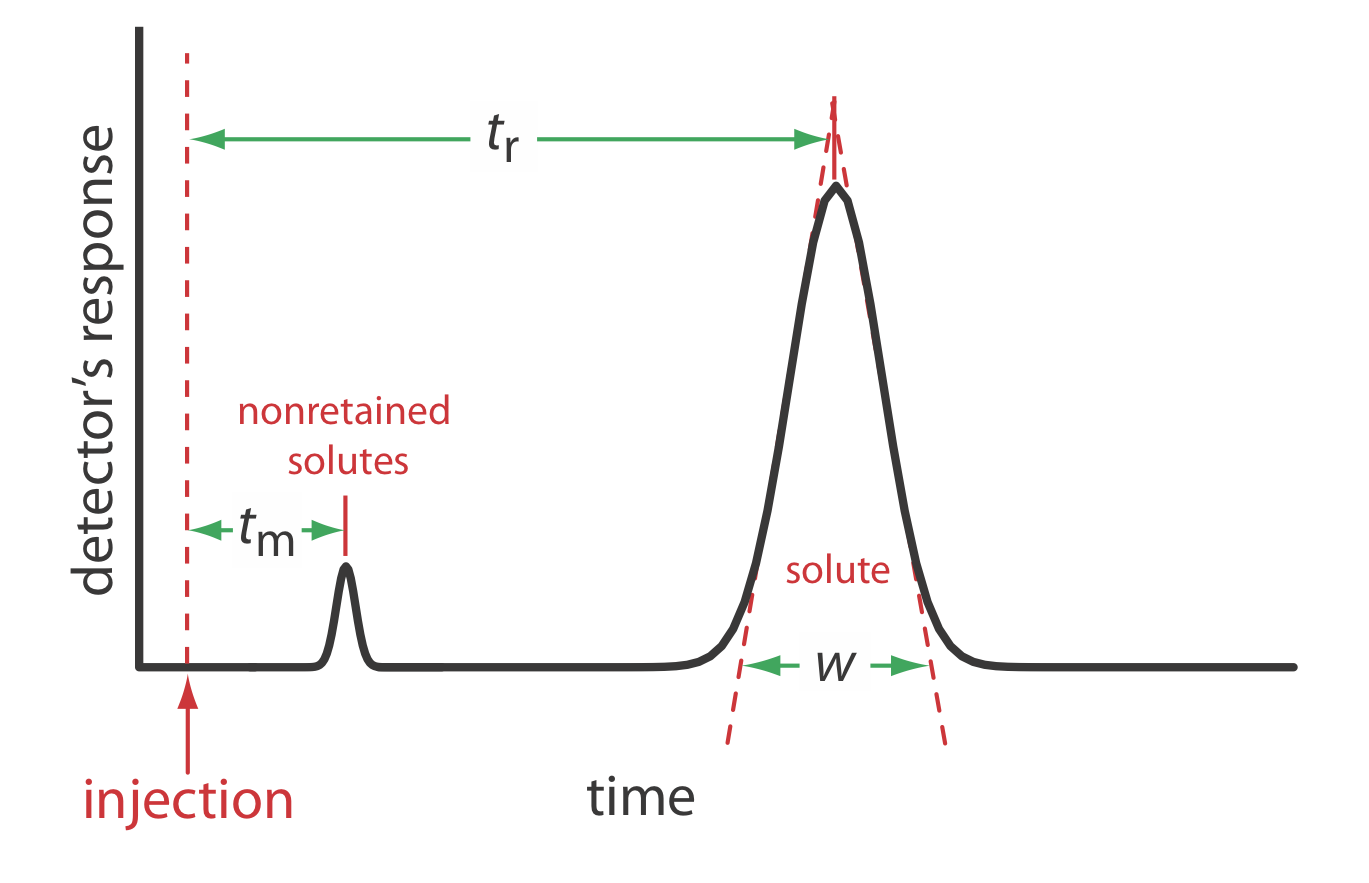
Además del pico del soluto, la Figura 12.2.4 también muestra un pequeño pico que eluye poco después de inyectar la muestra en la fase móvil. Este pico contiene todos los solutos no retenidos, que se mueven a través de la columna a la misma velocidad que la fase móvil. El tiempo requerido para eluir los solutos no retenidos se denomina tiempo vacío de la columna, t m.
Resolución Cromatográfica
El objetivo de la cromatografía es separar una mezcla en una serie de picos cromatográficos, cada uno de los cuales constituye un solo componente de la mezcla. La resolución entre dos picos cromatográficos, R AB, es una medida cuantitativa de su separación, y se define como
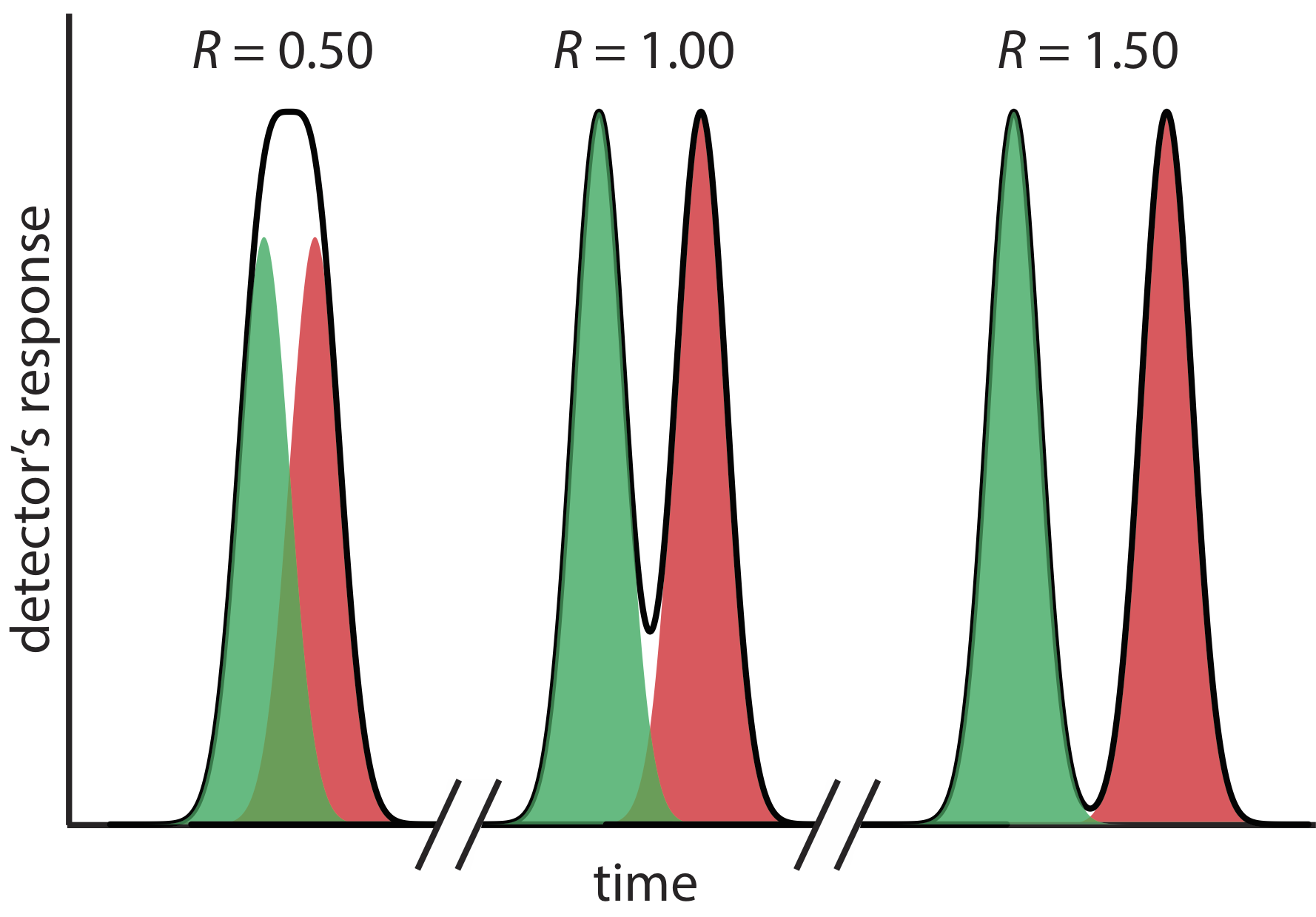
donde B es la elución posterior de los dos solutos. Como se muestra en la Figura 12.2.5 , la separación de dos picos cromatográficos mejora con un incremento en R AB. Si las áreas bajo los dos picos son idénticas, como es el caso en la Figura 12.2.5 , entonces una resolución de 1.50 corresponde a una superposición de solo 0.13% para los dos perfiles de elución. Debido a que la resolución es una medida cuantitativa del éxito de una separación, es una forma útil de determinar si un cambio en las condiciones experimentales conduce a una mejor separación.
En un análisis cromatográfico de aceite de limón un pico para limoneno tiene un tiempo de retención de 8.36 min con un ancho basal de 0.96 min. \(\gamma\)-Terpineno eluye a 9.54 min con un ancho basal de 0.64 min. ¿Cuál es la resolución entre los dos picos?
Solución
Usando la ecuación\ ref {12.1} encontramos que la resolución es
\[R_{A B}=\frac{2 \Delta t_{r}}{w_{B}+w_{A}}=\frac{2(9.54 \text{ min}-8.36 \text{ min})}{0.64 \text{ min}+0.96 \text{ min}}=1.48 \nonumber\]
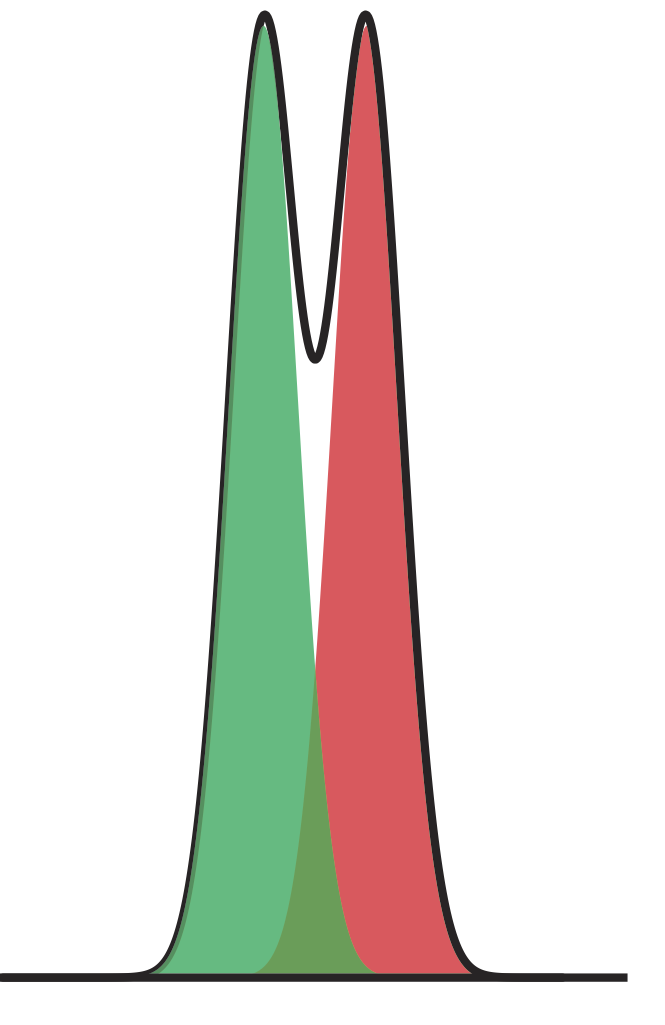
La Figura 12.2.6 muestra la separación de una mezcla de dos componentes. ¿Cuál es la resolución entre los dos componentes? Use una regla para medir\(\Delta t_\text{r}\), w A y w B en milímetros.
- Responder
-
Debido a que la relación entre el tiempo de elución y la distancia es proporcional\(\Delta t_\text{r}\), podemos medir, w A y w B usando una regla. Mis medidas son 8.5 mm para\(\Delta t_\text{r}\), y 12.0 mm cada una para w A y w B. Usando estos valores, la resolución es
\[R_{A B}=\frac{2 \Delta t_{t}}{w_{A}+w_{B}}=\frac{2(8.5 \text{ mm})}{12.0 \text{ mm}+12.0 \text{ mm}}=0.70 \nonumber\]
Sus medidas para\(\Delta t_\text{r}\), w A y w B dependerán del tamaño relativo de su monitor o impresión; sin embargo, su valor para la resolución debe ser similar a la respuesta anterior.
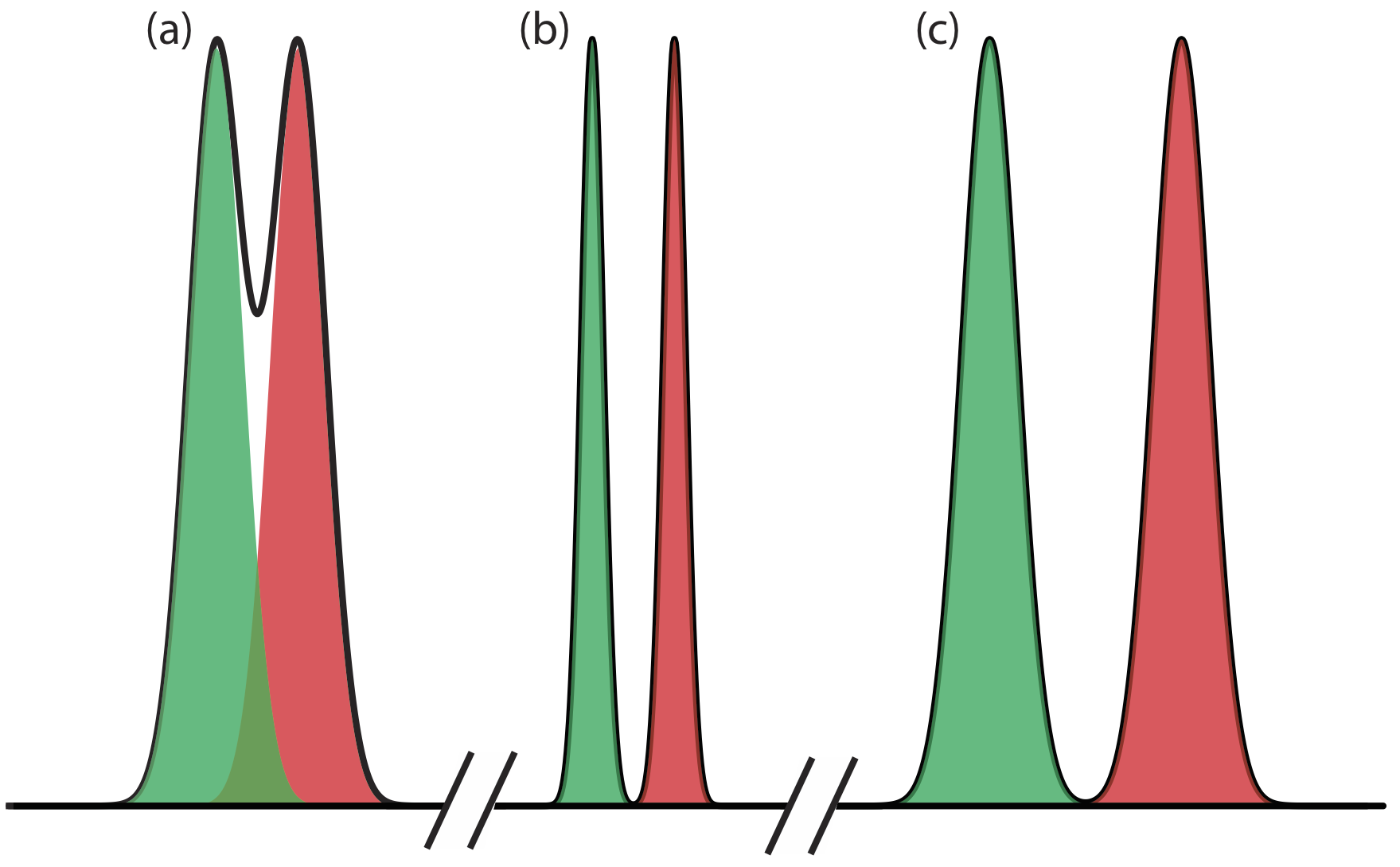
La ecuación\ ref {12.1} sugiere que podemos mejorar la resolución aumentando\(\Delta t_\text{r}\), o disminuyendo w A y w B (Figura 12.2.7 ). Para aumentar\(\Delta t_\text{r}\) podemos usar una de dos estrategias. Un enfoque es ajustar las condiciones de separación para que ambos solutos pasen menos tiempo en la fase móvil, es decir, aumentemos el factor de retención de cada soluto, lo que proporciona más tiempo para efectuar una separación. Un segundo enfoque es aumentar la selectividad ajustando las condiciones para que solo un soluto experimente un cambio significativo en su tiempo de retención. El ancho basal del pico de un soluto depende del movimiento de solutos dentro y entre la fase móvil y la fase estacionaria, y se rige por varios factores que colectivamente llamamos eficiencia de columna. Consideraremos cada uno de estos enfoques para mejorar la resolución con más detalle, pero primero debemos definir algunos términos.
Factor de retención de solutos
Supongamos que podemos describir la distribución de un soluto entre la fase móvil y la fase estacionaria usando la siguiente reacción de equilibrio
\[S_{\text{m}} \rightleftharpoons S_{\text{s}} \nonumber\]
donde S m es el soluto en la fase móvil y S s es el soluto en la fase estacionaria. Siguiendo el mismo enfoque que utilizamos en el Capítulo 7.7 para las extracciones líquido-líquido, la constante de equilibrio para esta reacción es un coeficiente de partición de equilibrio, K D.
\[K_{D}=\frac{\left[S_{\mathrm{s}}\right]}{\left[S_\text{m}\right]} \nonumber\]
Esto no es una suposición trivial. En esta sección estamos, en efecto, tratando el equilibrio del soluto entre la fase móvil y la fase estacionaria como si fuera idéntico al equilibrio en una extracción líquido-líquido. Podría cuestionarse si se trata de una suposición razonable. Hay una diferencia importante entre los dos experimentos que debemos considerar. En una extracción líquido-líquido, que se realiza en un embudo separador, las dos fases permanecen en contacto entre sí en todo momento, permitiendo un verdadero equilibrio. En cromatografía, sin embargo, la fase móvil está en constante movimiento. Un soluto que se mueve a la fase estacionaria desde la fase móvil se equilibrará de nuevo en una porción diferente de la fase móvil; esto no describe un verdadero equilibrio.
Entonces, preguntamos nuevamente: ¿Podemos tratar la distribución de un soluto entre la fase móvil y la fase estacionaria como un proceso de equilibrio? La respuesta es sí, si la velocidad de la fase móvil es lenta en relación con la cinética del movimiento del soluto de ida y vuelta entre las dos fases. En general, esta es una suposición razonable.
En ausencia de cualquier reacción de equilibrio adicional en la fase móvil o la fase estacionaria, K D es equivalente a la relación de distribución, D,
donde V s y V m son los volúmenes de la fase estacionaria y la fase móvil, respectivamente.
Una conservación de la masa requiere que los moles totales de soluto permanezcan constantes a lo largo de la separación; así, sabemos que la siguiente ecuación es cierta.
Resolviendo la Ecuación\ ref {12.3} para los moles de soluto en la fase estacionaria y sustituyendo en la Ecuación\ ref {12.2} nos deja con
\[D = \frac{\left\{(\text{mol S})_{\text{tot}} - (\text{mol S})_\text{m}\right\} / V_{\mathrm{s}}}{(\text{mol S})_{\mathrm{m}} / V_{\mathrm{m}}} \nonumber\]
Reordenando esta ecuación y resolviendo para la fracción de soluto en la fase móvil, f m, da
que es idéntico al resultado de una extracción líquido-líquido (ver Capítulo 7). Debido a que tal vez no sepamos los volúmenes exactos de la fase estacionaria y la fase móvil, simplificamos la Ecuación\ ref {12.4} dividiendo tanto el numerador como el denominador entre V m; así
donde k
\[k=D \times \frac{V_\text{s}}{V_\text{m}} \label{12.6}\]
es el factor de retención del soluto. Tenga en cuenta que cuanto mayor sea el factor de retención, más favorece la relación de distribución a la fase estacionaria, lo que lleva a un soluto retenido más fuertemente y a un mayor tiempo de retención.
Otros nombres (más antiguos) para el factor de retención son factor de capacidad, relación de capacidad y relación de partición, y a veces se le da el símbolo\(k^{\prime}\). Tenga esto en cuenta si está utilizando otros recursos. El factor de retención es el nombre aprobado del Libro de Oro de la IUPAC.
Podemos determinar un factor de retención de soluto a partir de un cromatograma midiendo el tiempo de vacío de la columna, t m, y el tiempo de retención del soluto, t r (ver Figura 12.2.4 ). Resolviendo la ecuación\ ref {12.5} para k, encontramos que
\[k=\frac{1-f_\text{m}}{f_\text{m}} \label{12.7}\]
Anteriormente definimos f m como la fracción de soluto en la fase móvil. Suponiendo una velocidad de fase móvil constante, también podemos definir f m como
\[f_\text{m}=\frac{\text { time spent in the mobile phase }}{\text { time spent in the stationary phase }}=\frac{t_\text{m}}{t_\text{r}} \nonumber\]
Sustituir de nuevo a la Ecuación\ ref {12.7} y reorganizar nos deja con
donde\(t_\text{r}^{\prime}\) está el tiempo de retención ajustado.
En un análisis cromatográfico de ácidos de bajo peso molecular, el ácido butírico eluye con un tiempo de retención de 7.63 min. El tiempo de vacío de la columna es de 0.31 min. Calcular el factor de retención para el ácido butírico.
Solución
\[k_{\mathrm{but}}=\frac{t_{\mathrm{r}}-t_{\mathrm{m}}}{t_{\mathrm{m}}}=\frac{7.63 \text{ min}-0.31 \text{ min}}{0.31 \text{ min}}=23.6 \nonumber\]
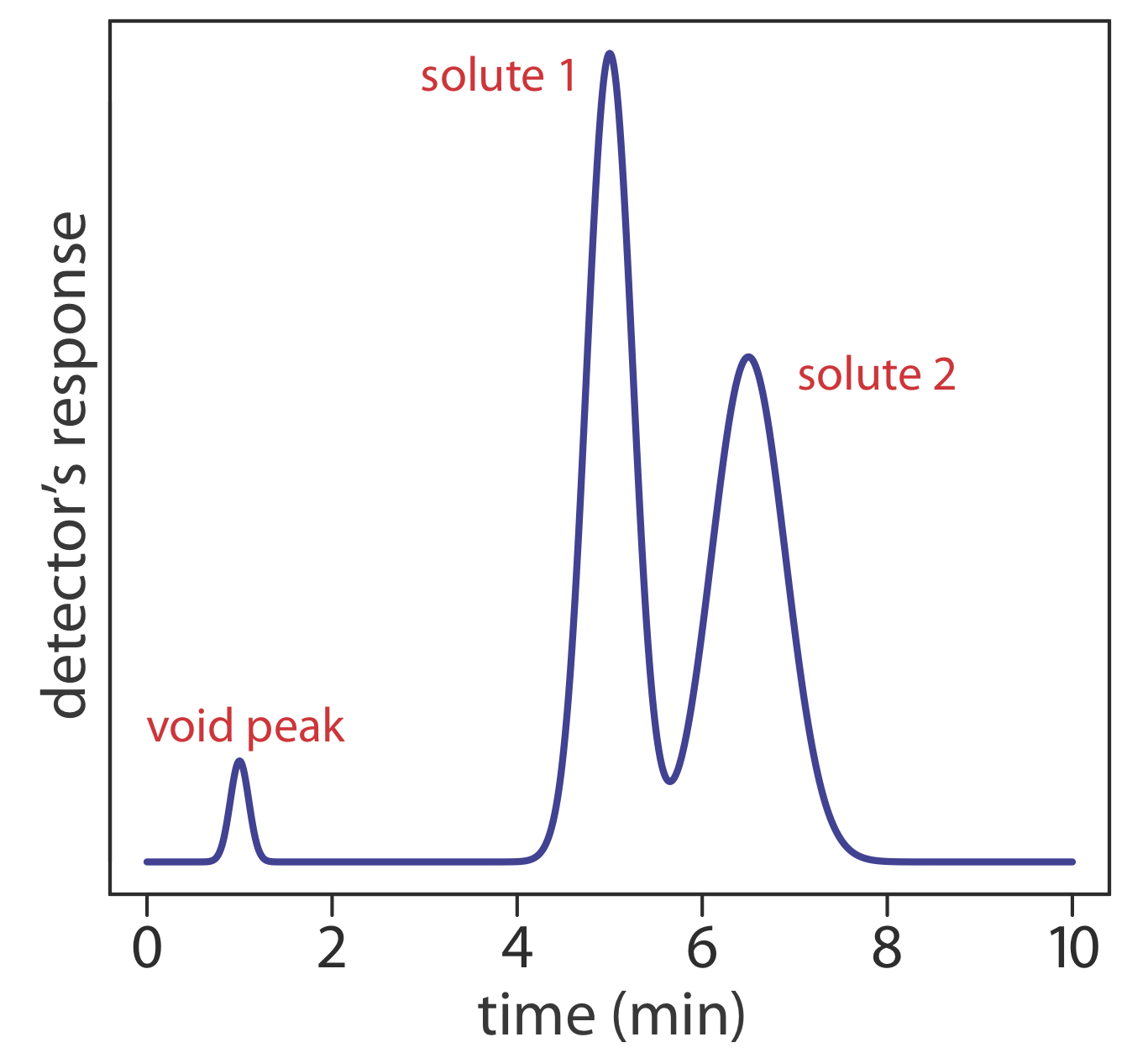
La Figura 12.2.8 es el cromatograma para una mezcla de dos componentes. Determinar el factor de retención para cada soluto asumiendo que la muestra fue inyectada en el tiempo t = 0.
- Responder
-
Debido a que la relación entre el tiempo de elución y la distancia es proporcional, podemos medir t m, t r,1 y t r,2 usando una regla. Mis medidas son 7.8 mm, 40.2 mm y 51.5 mm, respectivamente. Usando estos valores, los factores de retención para el soluto A y el soluto B son
\[k_{1}=\frac{t_{\mathrm{r} 1}-t_\text{m}}{t_\text{m}}=\frac{40.2 \text{ mm}-7.8 \text{ mm}}{7.8 \text{ mm}}=4.15 \nonumber\]
\[k_{2}=\frac{t_{\mathrm{r} 2}-t_\text{m}}{t_\text{m}}=\frac{51.5 \text{ mm}-7.8 \text{ mm}}{7.8 \text{ mm}}=5.60 \nonumber\]
Sus medidas para t m, t r,1 y t r,2 dependerán del tamaño relativo de su monitor o impresión; sin embargo, su valor para la resolución debe ser similar a la respuesta anterior.
Selectividad
La selectividad es una medida relativa de la retención de dos solutos, que definimos usando un factor de selectividad,\(\alpha\)
\[\alpha=\frac{k_{B}}{k_{A}}=\frac{t_{r, B}-t_{\mathrm{m}}}{t_{r, A}-t_{\mathrm{m}}} \label{12.9}\]
donde el soluto A tiene el menor tiempo de retención. Cuando dos solutos eluyen con idéntico tiempo de retención,\(\alpha = 1.00\); para todas las demás condiciones\(\alpha > 1.00\).
En el análisis cromatográfico para ácidos de bajo peso molecular descrito en el Ejemplo 12.2.2 , el tiempo de retención para el ácido isobutírico es de 5.98 min. ¿Cuál es el factor de selectividad para el ácido isobutírico y el ácido butírico?
Solución
Primero debemos calcular el factor de retención para el ácido isobutírico. Usando el tiempo vacío del Ejemplo 12.2.2 tenemos
\[k_{\mathrm{iso}}=\frac{t_{\mathrm{r}}-t_{\mathrm{m}}}{t_{\mathrm{m}}}=\frac{5.98 \text{ min}-0.31 \text{ min}}{0.31 \text{ min}}=18.3 \nonumber\]
El factor de selectividad, por lo tanto, es
\[\alpha=\frac{k_{\text {but }}}{k_{\text {iso }}}=\frac{23.6}{18.3}=1.29 \nonumber\]
Determinar el factor de selectividad para el cromatograma en el Ejercicio 12.2.2 .
- Responder
-
Usando los resultados del Ejercicio 12.2.2 , el factor de selectividad es
\[\alpha=\frac{k_{2}}{k_{1}}=\frac{5.60}{4.15}=1.35 \nonumber\]
Su respuesta puede diferir ligeramente debido a diferencias en sus valores para los dos factores de retención.
Eficiencia de Columna
Supongamos que inyectamos una muestra que tiene un solo componente. Por el momento inyectamos la muestra es una banda estrecha de ancho finito. A medida que la muestra pasa por la columna, el ancho de esta banda aumenta continuamente en un proceso que llamamos ensanchamiento de banda. La eficiencia de la columna es una medida cuantitativa de la extensión del ensanchamiento de banda.
Consulte la Figura 12.2.1 y la Figura 12.2.2 . Cuando inyectamos la muestra tiene un perfil de concentración uniforme, o rectangular con respecto a la distancia por la columna. A medida que pasa por la columna, la banda se ensancha y adquiere un perfil de concentración gaussiano.
En su modelo teórico original de cromatografía, Martin y Synge dividieron la columna cromatográfica en secciones discretas, a las que llamaron placas teóricas. Dentro de cada placa teórica existe un equilibrio entre el soluto presente en la fase estacionaria y el soluto presente en la fase móvil [Martin, A. J. P.; Synge, R. L. M. Biochem. J. 1941, 35, 1358—1366]. Describieron la eficiencia de la columna en términos del número de placas teóricas, N,
\[N=\frac{L}{H} \label{12.10}\]
donde L es la longitud de la columna y H es la altura de una placa teórica. Para cualquier columna dada, la eficiencia de la columna mejora —y los picos cromatográficos se vuelven más estrechos— cuando hay más placas teóricas.
Si asumimos que un pico cromatográfico tiene un perfil gaussiano, entonces la extensión del ensanchamiento de banda viene dada por la varianza o desviación estándar del pico. La altura de una placa teórica es la varianza del pico por unidad de longitud de la columna
\[H=\frac{\sigma^{2}}{L} \label{12.11}\]
donde la desviación estándar,\(\sigma\), tiene unidades de distancia. Debido a que los tiempos de retención y los anchos de pico generalmente se miden en segundos o minutos, es más conveniente expresar la desviación estándar en unidades de tiempo\(\tau\), dividiendo\(\sigma\) por la velocidad lineal promedio del soluto\(\overline{u}\), lo que equivale a dividir la distancia que recorre, L, por su tiempo de retención, t r.
\[\tau=\frac{\sigma}{\overline{u}}=\frac{\sigma t_{r}}{L} \label{12.12}\]
Para una forma de pico gaussiano, el ancho en la línea base, w, es cuatro veces su desviación estándar,\(\tau\).
Combinando la Ecuación\ ref {12.11}, Ecuación\ ref {12.12} y Ecuación\ ref {12.13} define la altura de una placa teórica en términos de los parámetros cromatográficos fácilmente medidos t r y w.
\[H=\frac{L w^{2}}{16 t_\text{r}^{2}} \label{12.14}\]
La ecuación de peinado\ ref {12.14} y la ecuación\ ref {12.10} da el número de placas teóricas.
\[N=16 \frac{t_{\mathrm{r}}^{2}}{w^{2}}=16\left(\frac{t_{\mathrm{r}}}{w}\right)^{2} \label{12.15}\]
Un análisis cromatográfico para el pesticida clorado Dieldrin da un pico con un tiempo de retención de 8.68 min y un ancho basal de 0.29 min. ¿Calcular el número de planchas teóricas? Dado que la columna tiene 2.0 m de largo, ¿cuál es la altura de una placa teórica en mm?
Solución
Usando la ecuación\ ref {12.15}, el número de placas teóricas es
\[N=16 \frac{t_{\mathrm{r}}^{2}}{w^{2}}=16 \times \frac{(8.68 \text{ min})^{2}}{(0.29 \text{ min})^{2}}=14300 \text{ plates} \nonumber\]
Resolviendo la ecuación\ ref {12.10} para H da la altura promedio de una placa teórica como
\[H=\frac{L}{N}=\frac{2.00 \text{ m}}{14300 \text{ plates}} \times \frac{1000 \text{ mm}}{\mathrm{m}}=0.14 \text{ mm} / \mathrm{plate} \nonumber\]
Para cada soluto en el cromatograma para el Ejercicio 12.2.2 , calcular el número de placas teóricas y la altura promedio de una placa teórica. La columna mide 0.5 m de largo.
- Responder
-
Debido a que la relación entre el tiempo de elución y la distancia es proporcional, podemos medir t r,1, t r,2, w 1 y w 2 usando una regla. Mis medidas son de 40.2 mm, 51.5 mm, 8.0 mm y 13.5 mm, respectivamente. Usando estos valores, el número de placas teóricas para cada soluto es
\[N_{1}=16 \frac{t_{r,1}^{2}}{w_{1}^{2}}=16 \times \frac{(40.2 \text{ mm})^{2}}{(8.0 \text{ mm})^{2}}=400 \text { theoretical plates } \nonumber\]
\[N_{2}=16 \frac{t_{r,2}^{2}}{w_{2}^{2}}=16 \times \frac{(51.5 \text{ mm})^{2}}{(13.5 \text{ mm})^{2}}=233 \text { theoretical plates } \nonumber\]
La altura de una placa teórica para cada soluto es
\[H_{1}=\frac{L}{N_{1}}=\frac{0.500 \text{ m}}{400 \text { plates }} \times \frac{1000 \text{ mm}}{\mathrm{m}}=1.2 \text{ mm} / \mathrm{plate} \nonumber\]
\[H_{2}=\frac{L}{N_{2}}=\frac{0.500 \text{ m}}{233 \text { plates }} \times \frac{1000 \text{ mm}}{\mathrm{m}}=2.15 \text{ mm} / \mathrm{plate} \nonumber\]
Sus medidas para t r,1, t r,2, w 1 y w 2 dependerán del tamaño relativo de su monitor o impresión; sin embargo, sus valores para N y para H deben ser similares a los respuesta anterior.
Es importante recordar que una placa teórica es un constructo artificial y que una columna cromatográfica no contiene placas físicas. De hecho, el número de placas teóricas depende tanto de las propiedades de la columna como del soluto. Como resultado, el número de placas teóricas para una columna puede variar de soluto a soluto.
Capacidad de pico
Una ventaja de mejorar la eficiencia de la columna es que podemos separar más solutos con resolución basal. Una estimación del número de solutos que podemos separar es
\[n_{c}=1+\frac{\sqrt{N}}{4} \ln \frac{V_{\max }}{V_{\min }} \label{12.16}\]
donde n c es la capacidad pico de la columna, y V min y V max son los volúmenes más pequeños y mayores de fase móvil en los que podemos eluir y detectar un soluto [Giddings, J. C. Unified Separation Ciencia, Wiley-Interscience: Nueva York, 1991]. Una columna con 10 000 placas teóricas, por ejemplo, puede resolver no más de
\[n_{c}=1+\frac{\sqrt{10000}}{4} \ln \frac{30 \mathrm{mL}}{1 \mathrm{mL}}=86 \text { solutes } \nonumber\]
si V min y V max son de 1 mL y 30 mL, respectivamente. Esta estimación proporciona un límite superior sobre el número de solutos y puede ayudarnos a excluir de consideración una columna que no tenga suficientes placas teóricas para separar una mezcla compleja. El hecho de que la capacidad máxima teórica de una columna sea mayor que el número de solutos, sin embargo, no significa que una separación sea factible. En la mayoría de las situaciones la capacidad pico práctica es menor que la capacidad máxima teórica debido a que las características de retención de algunos solutos son tan similares que una separación es imposible. Sin embargo, las columnas con placas más teóricas, o con un mayor rango de posibles volúmenes de elución, tienen más probabilidades de separar una mezcla compleja.
El volumen más pequeño que podemos usar es el volumen vacío de la columna. El mayor volumen está determinado ya sea por nuestra paciencia, el tiempo máximo de análisis que podemos tolerar, o por nuestra incapacidad para detectar solutos porque hay demasiada ampliación de banda.
Picos asimétricos
Nuestro tratamiento de la cromatografía en esta sección asume que un soluto eluye como un pico gaussiano simétrico, como el que se muestra en la Figura 12.2.4 . Este comportamiento ideal ocurre cuando el coeficiente de partición del soluto, K D
\[K_{\mathrm{D}}=\frac{[S_\text{s}]}{\left[S_\text{m}\right]} \nonumber\]
es lo mismo para todas las concentraciones de soluto. Si este no es el caso, entonces el pico cromatográfico tiene una forma de pico asimétrica similar a las que se muestran en la Figura 12.2.9 . El pico cromatográfico en la Figura 12.2.9 a es un ejemplo de cola de pico, que ocurre cuando algunos sitios en la fase estacionaria retienen el soluto más fuertemente que otros sitios. Figura 12.2.9 b, que es un ejemplo de frente de pico con mayor frecuencia es el resultado de sobrecargar la columna con muestra.
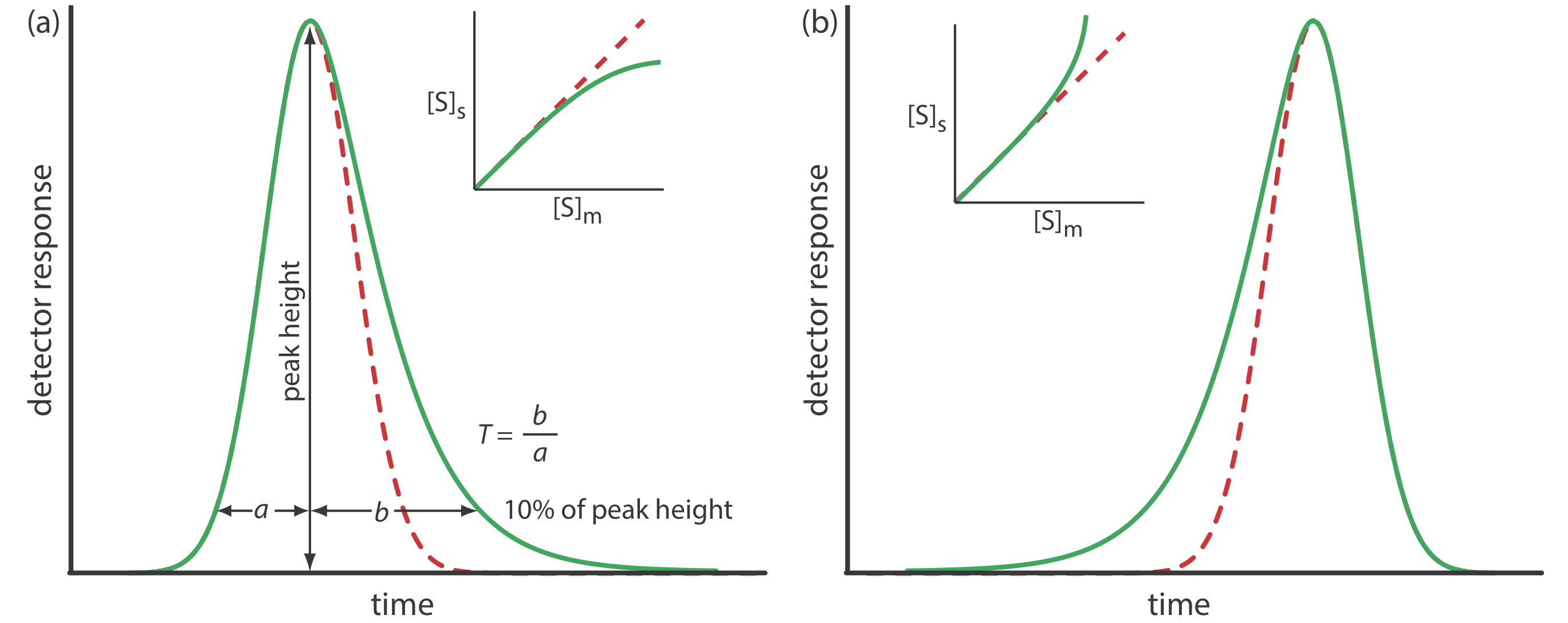
Como se muestra en la Figura 12.2.9 a, podemos reportar la asimetría de un pico dibujando una línea horizontal al 10% de la altura máxima del pico y midiendo la distancia desde cada lado del pico hasta una línea trazada verticalmente a través del máximo del pico. El factor de asimetría, T, se define como
\[T=\frac{b}{a} \nonumber\]
El número de placas teóricas para una forma de pico asimétrica es aproximadamente
\[N \approx \frac{41.7 \times \frac{t_{r}^{2}}{\left(w_{0.1}\right)^{2}}}{T+1.25}=\frac{41.7 \times \frac{t_{r}^{2}}{(a+b)^{2}}}{T+1.25} \nonumber\]
donde w 0.1 es el ancho al 10% de la altura del pico [Foley, J. P.; Dorsey, J. G. Anal. Chem. 1983, 55, 730—737].
Los picos asimétricos tienen menos placas teóricas, y cuanto más asimétrico es el pico, menor es el número de placas teóricas. Por ejemplo, la siguiente tabla da valores para N para un soluto eluyendo con un tiempo de retención de 10.0 min y un ancho de pico de 1.00 min.
| b | a | T | N |
|---|---|---|---|
|
0.5 |
0.5 |
1.00 | 1850 |
|
0.6 |
0.4 |
1.50 | 1520 |
|
0.7 |
0.3 |
2.33 | 1160 |
| 0.8 | 0.2 | 4.00 | 790 |