
8.5: Gases reales
- Page ID
- 69792
En lo que va de este capítulo, la mayoría de ustedes no han aprendido nada que no hayan aprendido en sus cursos de cálculo. Las matemáticas en este capítulo son en realidad bastante fáciles, pero sin embargo, es común que a los estudiantes les resulte muy difícil aplicar estas herramientas matemáticas a problemas reales en las ciencias físicas. Para que se sienta cómodo usando las matemáticas en química primero aprenderemos un poco sobre los gases y la termodinámica. Tener algunos antecedentes nos ayudará a usar las matemáticas en un contexto con el que nosotros (los químicos) podamos relacionarnos.
Ya hemos mencionado algunas variables termodinámicas, pero para poder hacer más conexiones entre química y matemáticas necesitamos introducir algunos conceptos que necesitamos para comenzar a discutir gases reales. Hablarás de estos conceptos con más profundidad en CHM 346. Una variable termodinámica importante que se utiliza para caracterizar el estado de un sistema es la energía interna (\(U\)). Pensemos en un contenedor que contenga un gas (e.g.\(O_2\)). La energía interna del sistema es la suma de las siguientes contribuciones:
- Energía cinética: La energía cinética es la energía que tienen las moléculas debido a sus movimientos. Se relaciona con su velocidad, y como se esperaba, aumenta con el aumento de la temperatura, ya que las moléculas se mueven más rápido.
- Energía vibratoria y rotacional: Las moléculas almacenan energía en sus enlaces. Como ya discutimos, los átomos vibran alrededor de su posición de equilibrio, y hay una energía asociada a estas vibraciones. La energía vibratoria de una molécula también depende de la temperatura, y del número de enlaces. Los gases atómicos (He, Ar, etc) no tienen energía vibratoria. Las moléculas también rotan, y hay energía almacenada en estos movimientos. Al igual que en el caso de las vibraciones, los gases atómicos no tienen aportes de las rotaciones. Aprenderás sobre la energía vibracional y rotacional en CHM 345.
- Energía potencial: Esta es la energía debida a las interacciones entre las moléculas que componen el gas. Los átomos (por ejemplo, Ar) también interactuarán si se acercan lo suficiente. La energía de las interacciones entre las moléculas depende obviamente de la naturaleza química de las moléculas. Sabemos que las moléculas polares interactuarán más fuertemente que las moléculas no polares, y los átomos con más electrones (por ejemplo, Ar) interactuarán más fuertemente que los átomos de bajo número atómico (por ejemplo, He). Para un gas dado, estas interacciones dependen de la distancia entre las moléculas.
Por simplicidad, nos concentraremos en los gases atómicos, donde las únicas contribuciones\(U\) son la energía cinética (que depende solo de la temperatura), y la energía potencial. Ya aprendiste sobre el modelo más simple utilizado para describir el comportamiento de los gases: el gas ideal (o perfecto). Aprendiste que hay dos supuestos detrás del modelo. Primero las partículas no tienen ningún tamaño, lo que significa que puedes juntarlas lo más cerca que quieras. En realidad, los átomos tienen un tamaño, y si intentas juntarlos con demasiada fuerza las nubes electrónicas se repelerán entre sí. La otra suposición es que las partículas no interactúan entre sí a ninguna distancia. En realidad, esto tiene sentido sólo a densidades muy bajas, cuando las moléculas están muy alejadas entre sí. Sin embargo, a medida que las moléculas se acercan, experimentan fuerzas atractivas que en muchos casos son tan fuertes que dan como resultado la formación de un líquido. Por supuesto si los empujamos demasiado cerca las fuerzas se vuelven repulsivas, pero hay un rango de distancias en el que dominan fuerzas atractivas. Esto tiene sentido para las moléculas polares, pero ¿qué pasa con los átomos (por ejemplo, Ar), que no tienen un momento dipolar permanente? Probablemente se enteró de las fuerzas dispersivas londinenses, sin las cuales nunca seríamos capaces de licuar un gas noble. Las fuerzas londinenses son más fuertes para los átomos que contienen más electrones, y es por eso que el punto de ebullición de Xe es mucho mayor que el punto de ebullición del Ne.
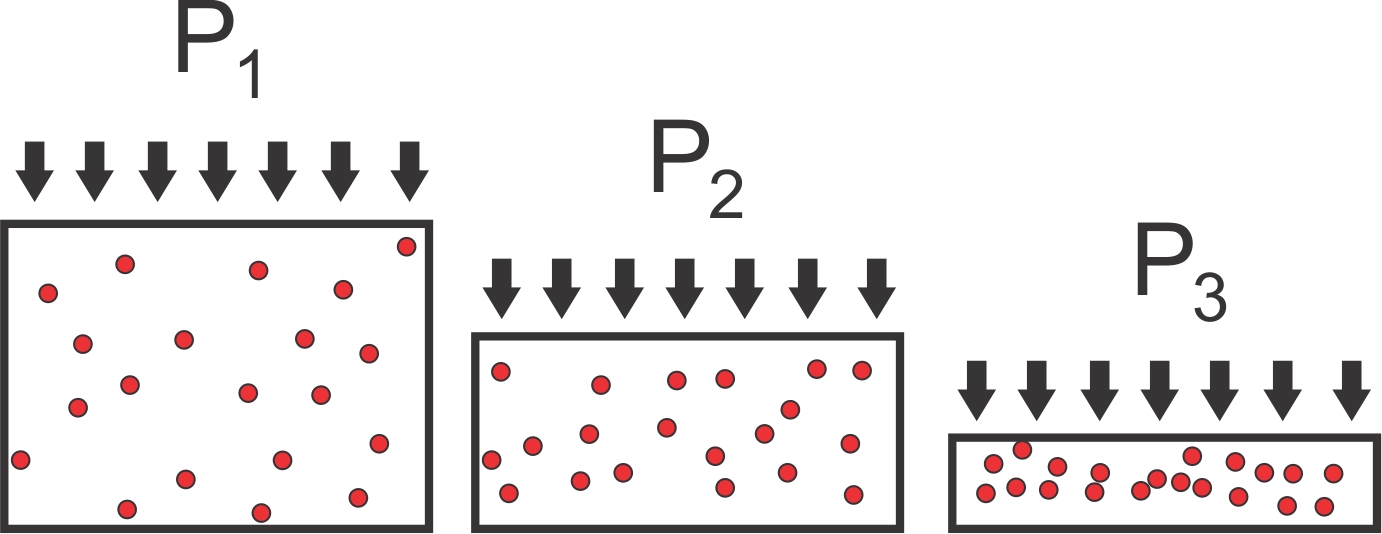
Con todo esto en mente, pensemos en lo que sucede con un gas ideal en las tres situaciones representadas en la Figura\(\PageIndex{1}\). Las densidades (moléculas por unidad de volumen) aumentan a medida que aumentamos la presión aplicada al recipiente. Supongamos que los tres contenedores están equilibrados a la misma temperatura, y pensemos en cómo se compara la energía interna entre los tres casos. Aún no tenemos ecuaciones, por lo que necesitamos pensar en términos de los conceptos que acabamos de discutir. Tenemos dos aportaciones en las que pensar. El término cinético debe ser el mismo en los tres contenedores porque la temperatura es la misma, y ese es el único factor que determina la velocidad de las moléculas. ¿Qué pasa con la energía potencial? Las partículas no tienen un tamaño, por lo que no hay fuerzas repulsivas que surjan si tratamos de acercarlas demasiado juntas. Tampoco tienen ningún tipo de interacciones atractivas, por lo que un gas ideal no almacena ninguna energía potencial. Acabamos de concluir que la energía interna para un gas ideal es igual a la energía cinética, y por lo tanto es una función de la temperatura del gas, pero no de su densidad. Ahora bien, sabemos que los gases ideales son representaciones simplificadas de gases reales, pero no existen. ¿En cuál de estas tres situaciones es más probable que un gas se comporte como un gas ideal? Claramente, cuando la densidad es baja, las moléculas están más alejadas, las interacciones entre ellas son más débiles y no necesitamos preocuparnos mucho por la energía potencial.
El modelo van der Waals
Volvamos a la ecuación de estado de un gas ideal\ ref {c2v:ideal}:
\[P=\frac{nRT}{V} \label{c2v:ideal}\]
Para mejorar nuestra descripción de los gases necesitamos tomar en cuenta los dos factores que el modelo de gas ideal descuida: el tamaño de las moléculas, y las interacciones entre ellas. El tamaño de las moléculas se puede tomar en cuenta asumiendo que el volumen que tienen que mover las moléculas no es realmente\(V\), sino\(V-nb\), donde\(nb\) está una medida del volumen que las moléculas ocupan ellas mismas. En este contexto\(b\) es una medida del volumen de una molécula, por lo que\(nb\) toma en cuenta el volumen de todas las moléculas presentes en el gas. Esta primera corrección da:
\[P=\frac{nRT}{V-nb}\label{calculus2v:eq:hardsphares}\]
que se conoce como el modelo de 'esferas duras'. Tenga en cuenta que aún no hemos introducido nada respecto a las interacciones, por lo que este modelo nos dice que podemos aumentar la densidad tanto como queramos sin cambiar la energía interna hasta llegar al punto en que las esferas se tocan entre sí. Debido a que son 'duras', la fuerza requerida para reducir aún más el volumen sería infinitamente grande (no muy diferente a empujar una bola de billar dentro de otra). Traducido en energía potencial, esto significa que la energía potencial saltará al infinito cuando la distancia entre el centro de las partículas sea igual a dos veces su radio. Esto es mejor que nada, pero no del todo realista. En realidad las moléculas no son completamente duras, y pueden ser empujadas unas contra otras un poco sin una fuerza infinita.
¿Qué pasa con las interacciones? Se discutió que a densidades moderadas, las interacciones atractivas dominan sobre las interacciones repulsivas. Para incorporar una corrección debido a interacciones atractivas necesitamos reconocer que la presión del gas necesita ser menor que la presión de un gas sin atracciones (como el modelo de esferas duras). La presión de un gas es una medida de las colisiones de las moléculas con las paredes del contenedor. Las fuerzas atractivas deben disminuir esta frecuencia, por lo que la presión resultante debe ser menor:
\[P=\frac{nRT}{V-nb}-C \nonumber\]
donde\(C\) será un término positivo, que tome en cuenta las interacciones atractivas. ¿De qué debería depender este término? Claramente sobre la naturaleza química de las moléculas, y debería ser mayor para átomos con más electrones (por ejemplo, Xe) que para átomos con menos electrones (por ejemplo, He). Además, debe depender de la densidad del gas (\(n/V\)), ya que las fuerzas atractivas son más fuertes cuanto más cerca están las moléculas. Van der Waals propuso la siguiente ecuación que satisface todo lo que acabamos de decir:
\[\label{c2v:eq:vdw} P=\frac{nRT}{V-nb}-a \left(\frac{n}{V}\right)^2\]
Consulta los siguientes ejemplos de constantes de van der Waals, y mira si entiendes cómo los valores de\(a\) y tienen\(b\) sentido en términos de los tamaños de las moléculas, y lo que sabes sobre las interacciones químicas de tus cursos generales de química. Preste atención a las unidades también.
| gas | \(a (L^2 bar/mol^{2})\) | \(b (L/mol)\) |
|---|---|---|
| Él | \ (a (L^2 bar/mol^ {2})\) ">0.035 | \ (b (L/mol)\) ">0.0237 |
| Ar | \ (a (L^2 bar/mol^ {2})\) ">1.35 | \ (b (L/mol)\) ">0.0320 |
| Kr | \ (a (L^2 bar/mol^ {2})\) ">2.349 | \ (b (L/mol)\) ">0.0398 |
| H\(_2\) | \ (a (L^2 bar/mol^ {2})\) ">0.248 | \ (b (L/mol)\) ">0.02661 |
| O\(_2\) | \ (a (L^2 bar/mol^ {2})\) ">1.378 | \ (b (L/mol)\) ">0.03183 |
| H\(_2\) O | \ (a (L^2 bar/mol^ {2})\) ">5.536 | \ (b (L/mol)\) ">0.03049 |
| CO\(_2\) | \ (a (L^2 bar/mol^ {2})\) ">3.64 | \ (b (L/mol)\) ">0.0427 |
Cabe destacar que el modelo van der Waals sigue siendo un modelo, es decir, no es la representación exacta de un gas real. Mejora muchas de las deficiencias de la ley real del gas, pero sigue siendo un modelo.
Volviendo a Figura\(\PageIndex{1}\), imagínese ahora que los contenedores están llenos de un gas van der Waals. Los tres estados tienen diferente energía interna ahora, porque la densidad de las moléculas es diferente, y eso cambia las fuerzas entre ellos. La energía potencial es cero si las moléculas están demasiado lejos para que cualquier fuerza atractiva o repulsiva sea significativa, o cuando las fuerzas atractivas y repulsivas se cancelan exactamente entre sí. Cuando las fuerzas atractivas dominan necesitaríamos trabajar para separar las moléculas, y la energía potencial es negativa. Cuando las fuerzas repulsivas dominan la energía potencial es positiva, y necesitaríamos trabajar para acercar las moléculas. Con todo esto en mente, ¿en cuál de las tres situaciones es menor la energía potencial? La respuesta es el contenedor número 3. Las moléculas están más cerca (pero no lo suficientemente cerca como para tocarse entre sí), por lo que las fuerzas atractivas son más fuertes que en el contenedor número 1. Las fuerzas atractivas bajan esa energía interna del sistema. Estos argumentos permiten trazar la energía potencial para un gas van der Waals en función de la distancia entre el centro de las moléculas:
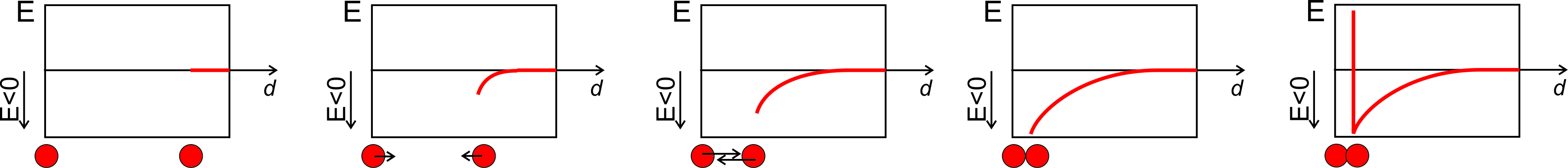
La energía potencial es cero cuando la distancia entre moléculas es mucho más larga que sus diámetros. Si comenzamos a disminuir la densidad se acercan, y las interacciones atractivas se vuelven significativas, bajando la energía interna. Esto continúa hasta que se tocan entre sí. Debido a que son 'esferas duras' no pueden penetrar entre sí en absoluto, y la energía potencial salta al infinito. Esto equivale a decir que la fuerza requerida para acercarlos es infinitamente grande.
Se discutió la diferencia entre un gas real y un modelo de un gas real. ¿Cómo se ve esta trama en la realidad y qué tan diferente es del modelo de van der Waals? A continuación se presentan ejemplos de diferentes gases. Observe que 'Ar\(_2\) 'no se refiere a un gas compuesto por moléculas de Ar\(_2\), sino a las interacciones entre dos átomos de Ar. Todos los gases en esta figura, como sabemos, son monoatómicos.
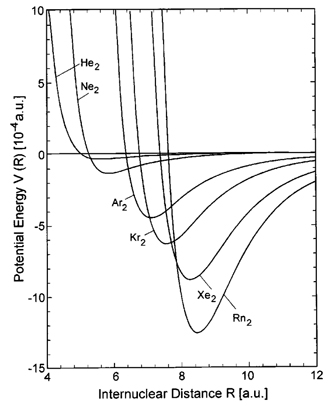
Hay algunas cosas que vale la pena señalar. Primero, los átomos con más electrones (mira una tabla periódica) muestran un pozo más profundo. Esto tiene sentido porque más electrones resultan en fuerzas londinenses más fuertes y atractivas. Además, los átomos con más electrones experimentan estas fuerzas atractivas a distancias más largas que los átomos con menos electrones. En todos los casos la energía potencial aumenta muy bruscamente cuando seguimos disminuyendo la distancia entre los átomos, pero la energía potencial no salta al infinito repentinamente, como en el caso del gas van der Waals. Esto significa que los átomos no son precisamente esferas duras. Como era de esperar, podemos acercar dos átomos de He mucho más que dos átomos de Rn antes de que veamos estas interacciones repulsivas porque los átomos de He son mucho más pequeños que los de Rn.
Isotermas de presión-volumen
Los científicos comenzaron a estudiar el comportamiento de los gases allá por el siglo XVI. Como se puede imaginar, contaban con insumos de laboratorio muy rudimentarios, y sus observaciones fueron en su mayoría cualitativas. Uno de los primeros estudios cuantitativos en química fue realizado por Robert Boyle, quien notó que el volumen y la presión de una cantidad fija de gas a temperatura constante cambian de acuerdo con la ley simple\(P\propto 1/V\), donde el símbolo “\(\propto\)” significa “proporcional a”. Esto es por supuesto cierto para un gas ideal, cuya ecuación de estado es\(P=nRT/V\), pero no para un gas real. La ley de Boyle predice que el volumen de un gas disminuye a medida que aumenta la presión a temperatura constante. Matemáticamente, esta curva se llama hipérbola (las hipérbolas son gráficas donde el producto\(xy\) es una constante), y físicamente llamamos a estas gráficas isotermas, porque representan el comportamiento a temperatura constante (iso significa igual en latín). En otras palabras, la isoterma para un gas ideal es una hipérbola, pero la isoterma para un gas real mostrará desviaciones de la forma hiperbólica.
Por supuesto, podemos ir al laboratorio y medir las isotermas para cualquier gas real en cualquier rango de temperaturas que queramos. Sabemos que siempre que las condiciones son tales que la densidad del gas es baja, esperamos que las interacciones sean insignificantes, y por lo tanto las isotermas deben estar muy cerca de las hipérbolas que Boyle describió en el siglo XIX. ¿Qué sucede cuando las interacciones son significativas? Algunas isotermas experimentales para CO\(_2\) se muestran en la Figura\(\PageIndex{4}\). A temperaturas más altas (e.g. 50\(^{\circ}\) C), la isoterma se acerca a la predicción de un gas ideal: podemos comprimir el volumen a pequeños volúmenes y la presión aumentará siguiendo la ley de Boyle. Las isotermas a temperaturas más altas estarán aún más cerca de las hipérbolas predichas por la ley de Boyle porque las interacciones se vuelven cada vez menos perceptibles.
¿Qué sucede a temperaturas más bajas? Consideremos la isoterma a 20\(^{\circ}\) C. Imagina que tienes un recipiente con un mol de CO\(_2\), y comienzas a reducir su volumen a temperatura constante. El volumen inicial se encuentra entre 0.5 y 0.6 L (punto A en la Figura\(\PageIndex{4}\)). A medida que comienzas a reducir el volumen la presión comienza a aumentar de acuerdo aproximado con la ley de Boyle, pero fíjate que se observan desviaciones importantes a medida que te acercas al punto B. Cuando la presión alcanza los 60 atm (punto C), el comportamiento del fluido se desvía mucho de lo predicho por las leyes del ideal gases. Sigues reduciendo el volumen del gas, pero la presión no solo no sube como predijo el bajo de Boyle, ¡sino que se mantiene constante por un tiempo (entre los puntos C y E)! ¿Qué está pasando? ¿Cómo podemos reducir el volumen de un gas sin aumentar la presión? La respuesta es que ya no estamos tratando con un gas puro: estamos licuando parte de él. A medida que pasamos de C a E, aumentamos la cantidad de líquido y reducimos la cantidad de gas, y la presión del sistema permanece constante siempre y cuando tengamos gas y líquido en equilibrio. Por supuesto, cuanto más líquido tengamos, menor volumen\(_2\) ocupa el CO.
Como mencionamos antes, no se supone que los gases ideales formen líquidos porque en principio, las moléculas que componen el gas no tienen ningún tamaño y no experimentan ninguna interacción con las otras moléculas en el contenedor. Para formar un líquido, las moléculas necesitan experimentar fuertes fuerzas atractivas, o de lo contrario los movimientos que experimentan debido a su energía térmica no les permitirían permanecer lo suficientemente cerca. Licuar un gas, por lo tanto, es una clara evidencia experimental de comportamiento no ideal y la existencia de interacciones atractivas entre moléculas.
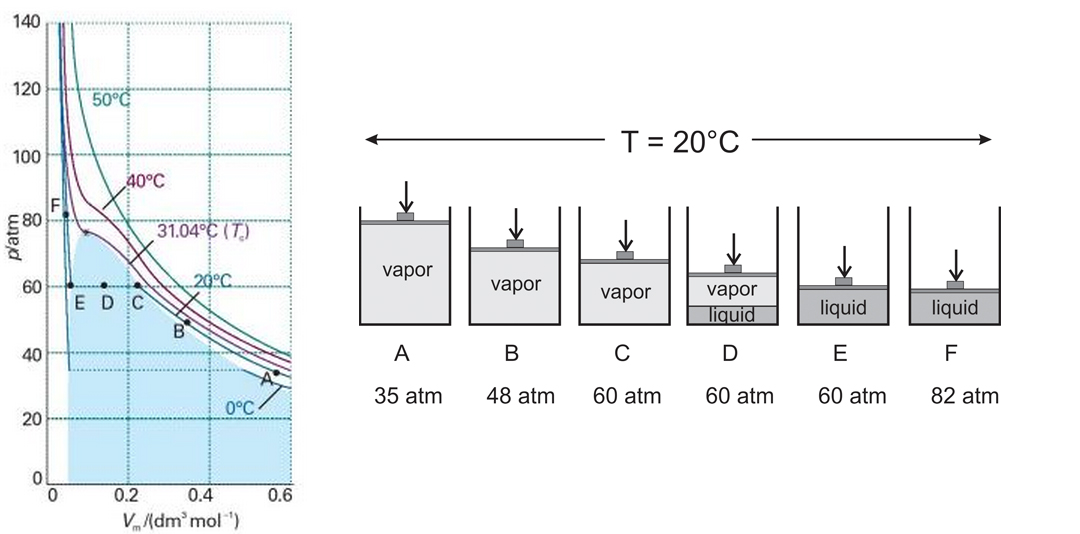
Volviendo a la Figura\(\PageIndex{4}\), cualquier punto de la línea horizontal CDE representa un estado donde coexisten líquido y gas. Nosotros llamamos al gas “vapor” en estas circunstancias, pero la distinción es más semántica que física. Cuando alcanzamos el punto E, todas las\(_2\) moléculas de CO son parte del líquido. Podemos seguir reduciendo el volumen, pero la presión del contenedor subirá mucho más dramáticamente que antes porque necesitaríamos ejercer una cantidad considerable de fuerza para empujar las moléculas de líquido más cerca entre sí. En términos más técnicos, los líquidos son mucho menos compresibles que los gases (ver la definición de compresibilidad en la página). El área resaltada en azul claro en la Figura\(\PageIndex{4}\) representa las condiciones bajo las cuales el CO\(_2\) puede existir en equilibrio entre las fases líquida y vapor. Por ejemplo, si realizamos el experimento a 0\(^{\circ}\) C, empezaríamos a ver las primeras gotas de líquido cuando la presión del contenedor llegue a\(\approx\) 35 atm. La presión se mantendrá constante a medida que continuemos reduciendo el volumen y formamos cada vez más líquido. Cuando no\(_2\) queda CO en fase vapor, reducir aún más el volumen requeriría que aumentemos drásticamente la presión del contenedor, ya que estaríamos comprimiendo un líquido, no un gas.
Observe que la longitud de la línea horizontal que representa la coexistencia de líquido y gas disminuye a medida que aumentamos la temperatura. Si tuviéramos que realizar el experimento a 30\(^{\circ}\) C (no mostrado), veríamos que el volumen al que vemos la primera gota de líquido no es demasiado diferente del volumen en el que dejamos de ver CO\(_2\) en la fase gaseosa. Licuar todo el gas requeriría un pequeño cambio de volumen, lo que significa que a esa temperatura y presión particulares, el volumen que ocupa el gas no es muy diferente del volumen que ocupa el líquido. Bastante extraño si lo piensas bien... un mol de un gas ideal ocupa 22.4L a temperatura ambiente, y un mol de agua líquida ocupa sólo 18 mL, casi mil veces menos. Si pensamos en términos de densidades, la densidad del agua a temperatura ambiente es de aproximadamente 1 g/mL, o 0.056 mol/mL. La densidad de un gas ideal a temperatura ambiente es\(n/V=P/RT\approx 4 \times 10^{-5}\) mol/L, nuevamente, alrededor de mil veces menos. Sin embargo, a 30\(^{\circ}\) C, y alrededor de 80 atm, la densidad de CO\(_2\) en estado líquido es casi la misma que la densidad de CO\(_2\) en la fase gaseosa. Si ponemos CO\(_2\) en una celda de alta presión, y aumentamos\(P\) a 80 atm, sería difícil para nosotros decir si el CO\(_2\) es líquido o gas. A presiones mucho más bajas, distinguir entre líquido y gas se hace mucho más evidente, como estamos acostumbrados a partir de nuestra experiencia diaria.
Comportamiento Crítico
Hay una isoterma particular donde la línea CDE de la figura [c2v:fig:isotermas] se reduce a un punto. En el caso del CO\(_2\), esta isoterma es la que medimos a 31.04\(^{\circ}\) C, y es tan única e importante que tiene un nombre especial: la 'isoterma crítica'. A temperaturas por debajo de la isoterma crítica, vemos que el gas se condensa para formar líquido, y que la presión del sistema permanece constante a medida que convertimos cada vez más gas en líquido. Cuanto menor es la temperatura, más diferentes son las densidades del vapor y del líquido. Esto es muy intuitivo para nosotros, porque es lo que estamos acostumbrados a ver con el líquido que mejor conocemos: el agua. En el caso del agua, necesitaríamos aumentar la presión a 218 atm y trabajar a 374\(^{\circ}\) C para perder nuestra capacidad de distinguir entre agua líquida y vapor. Las condiciones en la tierra están tan alejadas del punto crítico, que podemos distinguir claramente el agua líquida del vapor de sus densidades. Volviendo al CO\(_2\), a medida que aumentamos la temperatura a altas presiones (más de 60 atm), los estados líquido y vapor del fluido se vuelven cada vez más similares. Justo por debajo de la temperatura crítica difícilmente podemos distinguir qué es líquido y qué es vapor, y exactamente a la temperatura crítica, esa distinción se pierde. Por encima de la temperatura crítica nunca vemos una separación de fases, solo vemos un fluido que se vuelve más denso a medida que reducimos el volumen del contenedor. Observe que el punto crítico es un punto de inflexión en la isoterma crítica. Esto sucede a un volumen molar particular (para CO\(_2\)\(V_c = 0.094\) L/mol) y a una presión particular (para CO\(_2\)\(P_c = 72.9\) atm), lo que llamamos el volumen molar crítico y la presión crítica del fluido. Si queremos licuar CO\(_2\), necesitamos hacerlo a temperaturas por debajo de su temperatura crítica (31.04\(^{\circ}\) C). A temperaturas por encima de este valor el fluido siempre será un gas, ¡aunque podría ser un gas muy denso! Los químicos llaman a este estado 'fluido supercrítico' solo para diferenciarlo de un gas de baja densidad como el CO\(_2\) a 1 atm. De nuevo, para darte una idea, un mol de CO\(_2\) ocupa alrededor de 25L a 1 atm y 40\(^{\circ}\) C, y lo llamamos gas sin pensarlo dos veces. De la figura [c2v:fig:isotermas], a 80 atm y 40\(^{\circ}\) C un mol de CO\(_2\) ocupa aproximadamente 0.15L, aproximadamente 170 veces menos que el gas que estamos acostumbrados a ver a 1 atm. Se trata de un fluido muy denso, pero técnicamente no es un líquido porque estamos por encima de la temperatura crítica. En su lugar, usamos el término fluido supercrítico. Resulta que el CO supercrítico\(_2\) es mucho más que una curiosidad. Se utiliza como solvente para muchos procesos industriales, incluyendo la descafeinación de café y la limpieza en seco.
De nuestra discusión anterior, es claro que los gases ideales no muestran un comportamiento crítico. Nuevamente, los gases ideales no existen, por lo que cuando decimos que los gases ideales no muestran un comportamiento crítico solo estamos diciendo que 1) los gases muestran un comportamiento crítico en condiciones de temperatura, presión y volumen molar que están muy lejos de las condiciones donde la ecuación simple\(PV=nRT\) describe el comportamiento de el gas y que 2) si queremos describir un gas cercano al punto crítico necesitamos una ecuación de estado que sea consistente con el comportamiento crítico. Si trazamos las isotermas de un gas ideal (\(P = nRT/V\)) obtendremos hipérbolas a cualquier temperatura. Nuevamente, esto funciona bien con gases a densidades muy bajas, pero debido a que el modelo no incluye interacciones, no puede describir las isotermas en o alrededor del punto crítico.
¿El modelo de las esferas duras de la ecuación\ ref {calculus2v:eq:hardsphares} es consistente con la existencia del punto crítico? Para responder a esta pregunta, podríamos trazar muchas isotermas de acuerdo con esta ecuación y ver si el modelo da una isoterma que tiene un punto de inflexión como la que se muestra en la Figura\(\PageIndex{4}\). Una vez más, hay que tener en cuenta que la cifra contiene los datos que medimos experimentalmente, que es lo que realmente\(_2\) está haciendo el CO en la naturaleza. Las ecuaciones de estado por definición son modelos que pretenden describir el sistema lo más cerca posible, pero son por definición simplificaciones del comportamiento real. Volviendo al modelo de esferas duras, puedes trazar tantas isotermas como quieras, pero verás que ninguna de ellas muestra un punto de inflexión. Esto es bastante obvio a partir de la ecuación, ya que las isotermas son básicamente las mismas que obtendríamos con la ecuación de gas ideal, pero simplemente desplazadas en el\(x-\) eje por la cantidad\(b\). Físicamente hablando, no es sorprendente porque el modelo de esferas duras no contiene ningún parámetro que explique las interacciones atractivas entre moléculas, y eso es lo que necesitamos para describir el comportamiento crítico.
Sabemos que la ecuación de van der Waals es la ecuación más simple que introduce un término para dar cuenta de fuerzas atractivas (Ecuación\ ref {c2v:eq:vdw}), por lo que es probable que su ecuación pueda ser consistente con el comportamiento crítico. Discutamos a qué nos referimos con esto con más detalle. Nuevamente, los gases van der Waals no existen en la naturaleza. Son una construcción teórica donde pensamos en las moléculas como esferas duras con energía cinética que interactúan entre sí de manera que la interacción promedio entre dos moléculas orientadas aleatoriamente es inversamente proporcional a la inversa de la sexta potencia de la distancia entre ellas. Si pudiéramos crear un gas cuyas moléculas siguieran estas leyes físicas exactas, el gas se comportaría exactamente como predice la ecuación de van der Waals. Entonces, ahora nos preguntamos: ¿ese gas mostraría una isoterma con un punto de inflexión como el que se muestra para el caso del CO real\(_2\)? La respuesta es sí, y las constantes críticas (\(P_c,T_c\)y\(V_c\)) dependen de los valores de los parámetros de van der Waals,\(a\) y\(b\).
Ahora bien, el hecho de que el modelo de van der Waals prediga el comportamiento crítico no significa en absoluto que describa bien toda la isoterma. Si trazas la ecuación de van der Waals a diferentes temperaturas verás que este modelo no predice la\(P-V\) parte “plana” de la curva, donde coexisten el líquido y el gas. Esto no es sorprendente, ya que el tratamiento de las interacciones atractivas en el modelo de van der Waals es demasiado sencillo para describir el estado líquido. De hecho, verás que la ecuación de van der Waals predice que la derivada\(\left(\frac{\partial P}{\partial V} \right)_{T,n}\) es positiva en ciertas regiones de la isoterma, lo que por supuesto no tiene ningún sentido físico. Comprimir el gas nunca bajará la presión como predice el gas van der Waals, por lo que podemos ver claramente cómo falla el modelo cuando las fuerzas atractivas son importantes son complejas. En cualquier caso, es bastante impresionante ver cómo una ecuación tan simple predice un comportamiento tan complejo como el punto crítico.