
8.2: Diagramas de fases de sustancias puras
- Page ID
- 77842

Un diagrama de fases es un mapa bidimensional que muestra qué fase o fases pueden existir en un estado de equilibrio bajo condiciones dadas. En este capítulo se describen diagramas de fase presión-volumen y presión-temperatura para una sola sustancia, y el capítulo 13 describirá numerosos tipos de diagramas de fase para sistemas multicomponentes.
8.2.1 Características de los diagramas de fase
Los diagramas de fase bidimensionales para un sistema de una sola sustancia se pueden generar como proyecciones de una superficie tridimensional en un sistema de coordenadas con ejes cartesianos\(p\),\(V/n\), y\(T\). Un punto en la superficie tridimensional corresponde a una combinación físicamente realizable de valores, para un estado de equilibrio del sistema que contiene una cantidad total\(n\) de la sustancia, de las variables\(p\),\(V/n\), y\(T\).
Los conceptos necesarios para interpretar diagramas de fases de una sola sustancia se ilustrarán con dióxido de carbono.
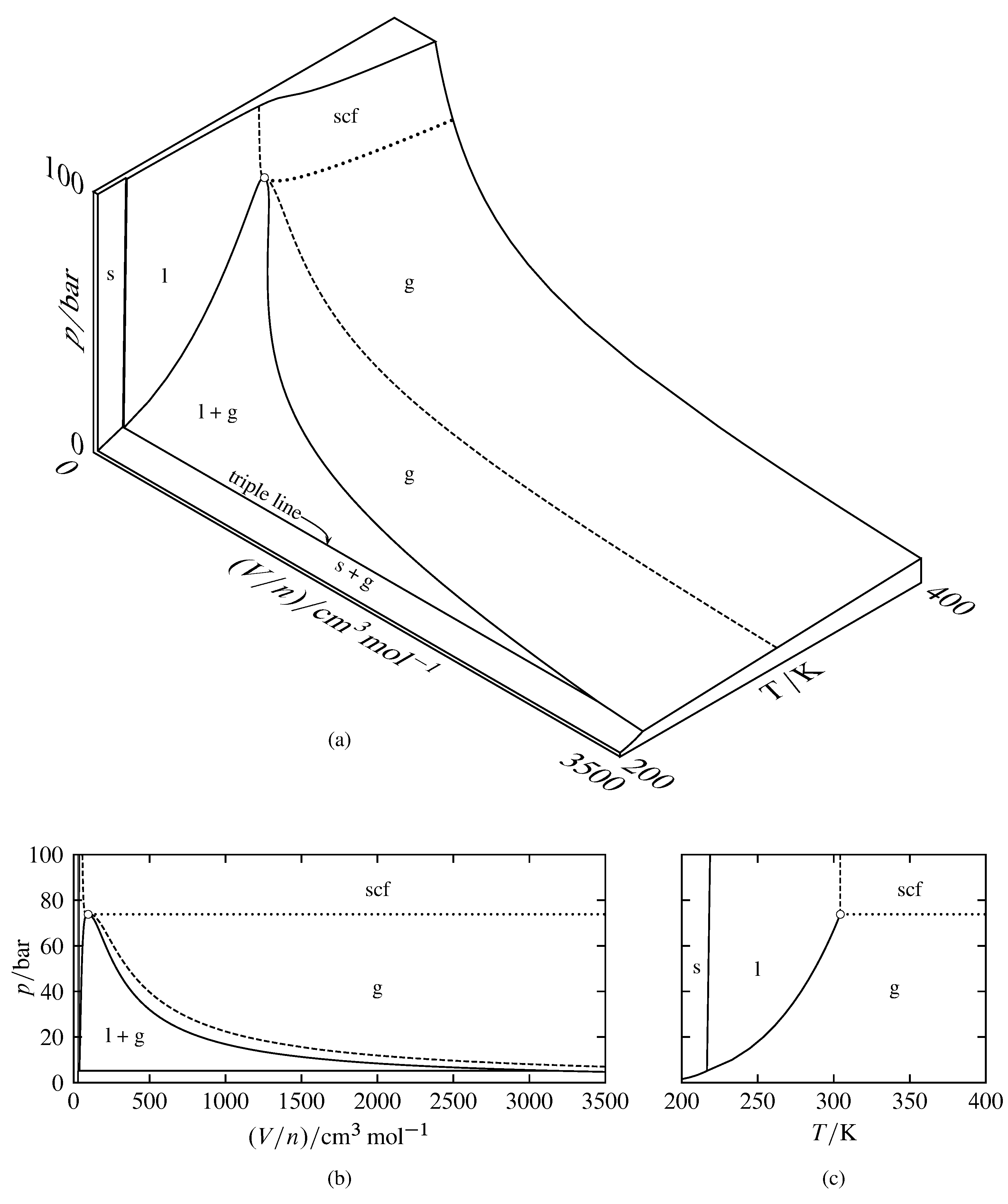
Figura 8.2 Relaciones entre\(p\)\(V/n\), y\(T\) para el dióxido de carbono (basado en datos en NIST Chemistry WebBook y en S. Angus, B. Armstrong, y K. M. de Reuck, Tablas Termodinámicas Internacionales del Estado Fluido, Vol. 3, Dióxido de Carbono, Pergamon Press, Oxford, 1976). Las áreas se etiquetan con la fase o fases estables (scf significa fluido supercrítico). El círculo abierto indica el punto crítico.
(a) Tridimensional\(p\) —\((V/n)\) —\(T\) superficie. La curva discontinua es la isoterma crítica en\(T=304.21\K\), y la curva punteada es una porción de la isobarra crítica en\(p=73.8\br\).
(b) Diagrama de fases presión-volumen (proyección de la superficie sobre el\((V/n)\) plano\(p\) -).
(c) Diagrama de fases presión-temperatura (proyección de la superficie sobre el\(T\) plano\(p\) -).
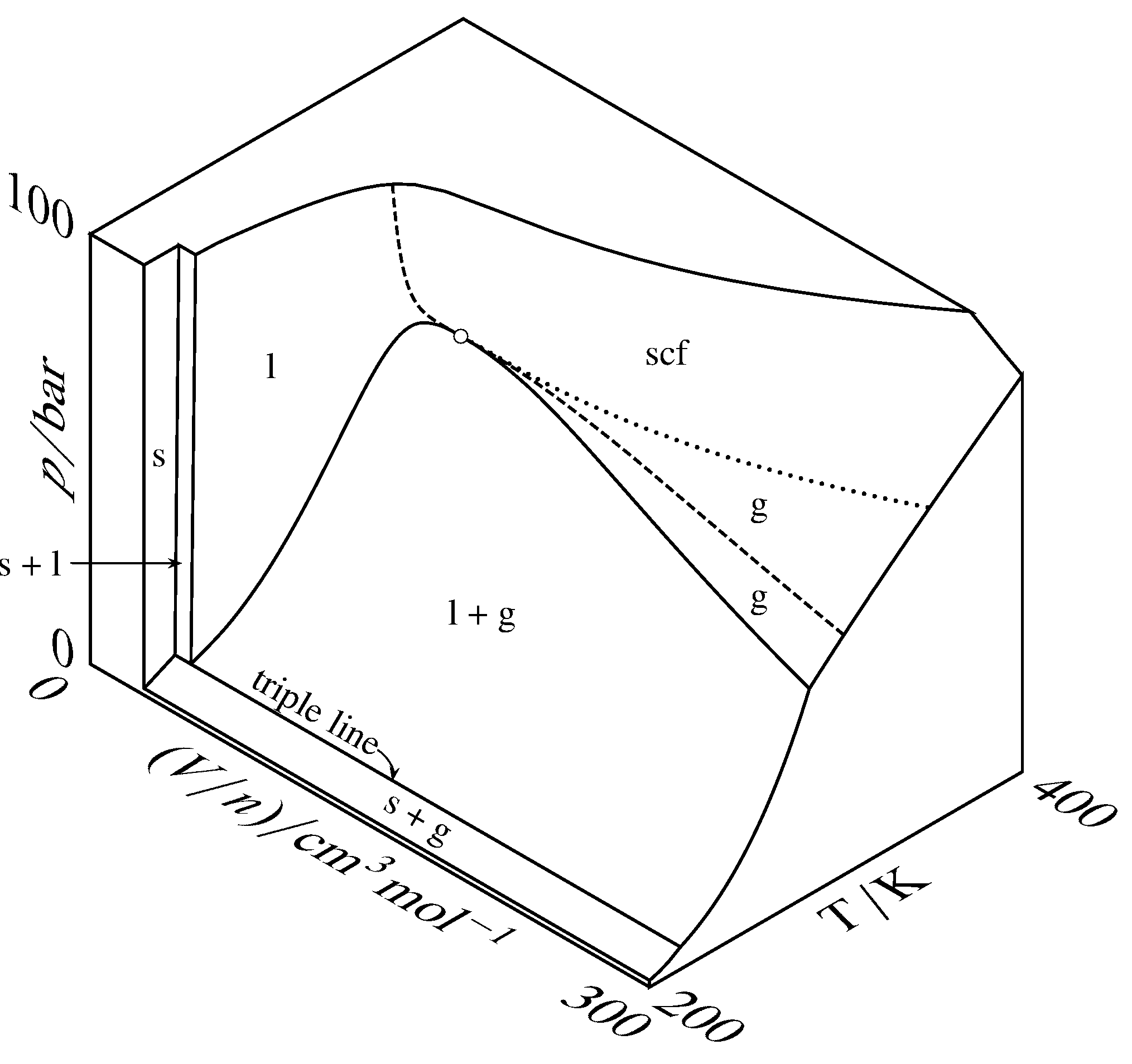
Figura 8.3\(T\) Superficie tridimensional\(p\) —\((V/n)\) — para CO\(_2\), magnificada a lo largo del\(V/n\) eje en comparación con la Fig. 8.2. El círculo abierto es el punto crítico, la curva discontinua es la isoterma crítica y la curva punteada es una porción de la isobarra crítica.
Las superficies tridimensionales para el dióxido de carbono se muestran a dos escalas diferentes en la Fig. 8.2 y en la Fig. 8.3. En estas figuras, algunas áreas de la superficie están etiquetadas con un solo estado físico: sólido, líquido, gas o fluido supercrítico. Un punto en una de estas áreas corresponde a un estado de equilibrio del sistema que contiene una sola fase del estado físico etiquetado. La forma de la superficie en esta área monofásica da la ecuación de estado de la fase (es decir, la dependencia de una de las variables de las otras dos). Un punto en un área etiquetada con dos estados físicos corresponde a dos fases coexistentes. La línea triple es el lugar de puntos para todos los sistemas de equilibrio posibles de tres fases coexistentes, que en este caso son sólido, líquido y gas. Un punto en la línea triple también puede corresponder a solo una o dos fases.
Las proyecciones bidimensionales mostradas en las figuras 8.2 (b) y 8.2 (c) son diagramas de fase presión-volumen y presión-temperatura. Debido a que todas las fases de un sistema de equilibrio multifásico tienen la misma temperatura y presión (suponiendo que no hay restricciones como particiones adiabáticas internas), la proyección de cada área bifásica sobre el diagrama de presión-temperatura es una curva, llamada curva de coexistencia o límite de fase, y la proyección de la línea triple es un punto, llamado punto triple.
¿Cómo podemos usar un diagrama de fases? Los dos ejes representan valores de dos variables independientes, tales como\(p\) y\(V/n\) o\(p\) y\(T\). Para valores dados de estas variables, colocamos un punto en el diagrama en la intersección de las coordenadas correspondientes; este es el punto del sistema. Entonces dependiendo de si el punto del sistema cae en un área o en una curva de coexistencia, el diagrama nos indica el número y tipos de fases que pueden estar presentes en el sistema de equilibrio.
Si el punto del sistema cae dentro de un área etiquetada con el estado físico de una sola fase, solo ese tipo de fase puede estar presente en el sistema de equilibrio. Un sistema que contiene una sustancia pura en una sola fase es bivariante (\(F = 3 - 1 = 2\)), por lo que podemos variar dos propiedades intensivas independientemente. Es decir, el punto del sistema puede moverse independientemente a lo largo de dos coordenadas (\(p\)\(p\)y\(V/n\), o y\(T\)) y aún permanecer en el área monofásica del diagrama de fases. Cuando\(V\) y\(n\) se refieren a una sola fase, la variable\(V/n\) es el volumen molar\(V\m\) en la fase.
Si el punto del sistema cae en un área del diagrama de fases presión-volumen etiquetado con símbolos para dos fases, estas dos fases coexisten en equilibrio. Las fases tienen la misma presión y diferentes volúmenes molares. Para encontrar los volúmenes molares de las fases individuales, dibujamos una línea horizontal de presión constante, llamada línea de unión, a través del punto del sistema y extendiéndose de un borde de la zona al otro. La posición horizontal de cada extremo de la línea de unión, donde termina en el límite con un área monofásica, da el volumen molar en esa fase en el sistema bifásico. Para ver un ejemplo de una línea de empate, véase la Fig. 8.9.
La línea triple en el diagrama presión-volumen representa el rango de valores\(V/n\) en los que tres fases (sólido, líquido y gas) pueden coexistir en equilibrio.
El helio es la única sustancia que carece de una línea triple sólido-líquido-gas. Cuando se enfría a un sistema que contiene el líquido y gas coexistentes de\({}^4\) He\(2.17\K\), se alcanza un punto triple en el que la tercera fase es un líquido llamado He-II, que tiene la propiedad única de superfluidez. Es sólo a altas presiones (\(10\br\)o mayores) que puede existir helio sólido.
Un sistema trifásico de un componente es invariante (\(F = 3 - 3 = 0\)); solo hay una temperatura (la temperatura de triple punto\(T\subs{tp}\)) y una presión (la presión de triple punto\(p\subs{tp}\)) a la que pueden coexistir las tres fases. Los valores de\(T\subs{tp}\) y\(p\subs{tp}\) son únicos para cada sustancia, y se muestran por la posición del punto triple en el diagrama de fases presión-temperatura. Los volúmenes molares en las tres fases coexistentes están dados por los valores de\(V/n\) en los tres puntos del diagrama presión-volumen donde la línea triple toca un área monofásica. Estos puntos están en los dos extremos y una posición intermedia de la triple línea. Si el punto del sistema está en cualquiera de los extremos de la línea triple, solo\(p\subs{tp}\) puede estar presente la fase del volumen molar correspondiente a temperatura\(T\subs{tp}\) y presión. Cuando el punto del sistema está en la línea triple en cualquier lugar entre los dos extremos, pueden estar presentes dos o tres fases. Si el punto del sistema está en la posición de la línea triple correspondiente a la fase de volumen molar intermedio, podría haber solo esa fase presente.
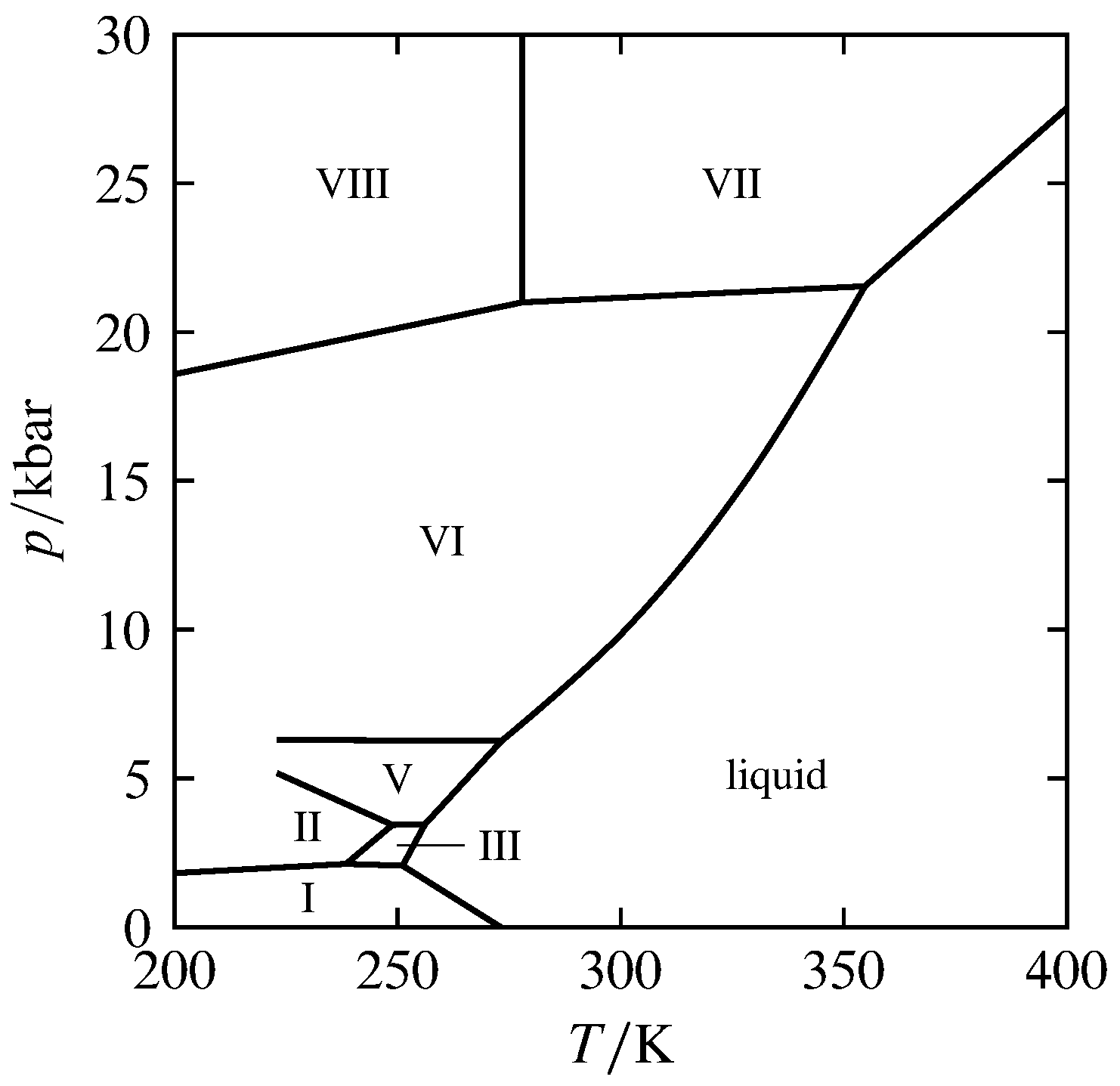
Figura 8.4 Diagrama de fases presión-temperatura de alta presión de H\(_2\) O (basado en datos de D. Eisenberg y W. Kauzmann, The Structure and Properties of Water, Oxford University Press, Nueva York, 1969, Tabla 3.5, y Carl W. F. T. Pistorius et al, J. Chem. Phys. , 38, 600—602, 1963). Los números romanos designan siete formas de hielo.
A altas presiones, una sustancia puede tener puntos triples adicionales para dos fases sólidas y la líquida, o para tres fases sólidas. Esto se ilustra mediante el diagrama de fases presión-temperatura de H\(_2\) O en la figura 8.4, que se extiende hasta presiones de hasta\(30\units{kbar}\). (En esta escala, la curva de coexistencia líquido-gas se encuentra demasiado cerca del eje horizontal para ser visible). El diagrama muestra siete fases sólidas diferentes de H\(_2\) O que difieren en la estructura cristalina y designadas hielo I, hielo II, y así sucesivamente. El hielo I es la forma ordinaria de hielo, estable debajo\(2\br\). En el diagrama hay cuatro puntos triples para dos sólidos y el líquido y tres puntos triples para tres sólidos. Cada punto triple es invariante. Observe cómo H\(_2\) O puede existir como hielo sólido VI o hielo VII por encima de su punto de fusión estándar de\(273\K\) si la presión es lo suficientemente alta (“hielo caliente”).
8.2.2 Equilibrio bifásico
Un sistema que contiene dos fases de una sustancia pura en equilibrio es univariante. Ambas fases tienen los mismos valores de\(T\) y de\(p\), pero estos valores no son independientes debido al requisito de que las fases tengan potenciales químicos iguales. Podemos variar solo una variable intensiva de una sustancia pura (tal como\(T\) o\(p\)) independientemente mientras que dos fases coexisten en equilibrio.
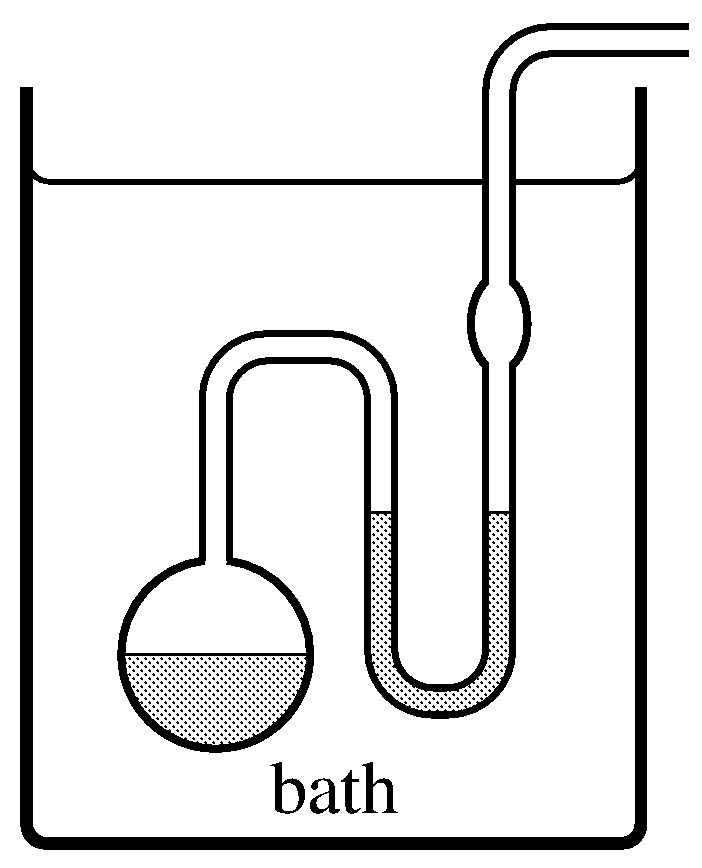
Figura 8.5 Un isoteniscopio. El líquido a investigar se coloca en el recipiente y tubo en U, como lo indica el sombreado, y se mantiene a una temperatura fija en el baño. La presión en el tubo lateral se reduce hasta que el líquido hierve suavemente y su vapor barre el aire. La presión se ajusta hasta que el nivel de líquido sea el mismo en ambas extremidades del tubo en U; la presión de vapor del líquido es entonces igual a la presión en el tubo lateral, que se puede medir con un manómetro.
A una temperatura dada, la presión a la que el sólido y el gas o el líquido y el gas están en equilibrio se denomina presión de vapor o presión de vapor de saturación del sólido o líquido. La presión de vapor de un sólido a veces se llama presión de sublimación. Podemos medir la presión de vapor de un líquido a una temperatura fija con un simple dispositivo llamado isoteniscopio (Fig. 8.5).
En un sistema de más de una sustancia, la presión de vapor puede referirse a la presión parcial de una sustancia en una mezcla gaseosa equilibrada con un sólido o líquido de esa sustancia. El efecto de la presión total sobre la presión de vapor se discutirá en la Sec. 12.8.1. Este libro electrónico se refiere a la presión de vapor de saturación de un líquido cuando es necesario indicar que son las fases de líquido puro y gas puro las que están en equilibrio a la misma presión.
A una presión dada, el punto de fusión o punto de congelación es la temperatura a la que el sólido y el líquido están en equilibrio, el punto de ebullición o temperatura de saturación es la temperatura a la que el líquido y el gas están en equilibrio, y la sublimación temperatura o punto de sublimación es la temperatura a la que el sólido y el gas están en equilibrio.
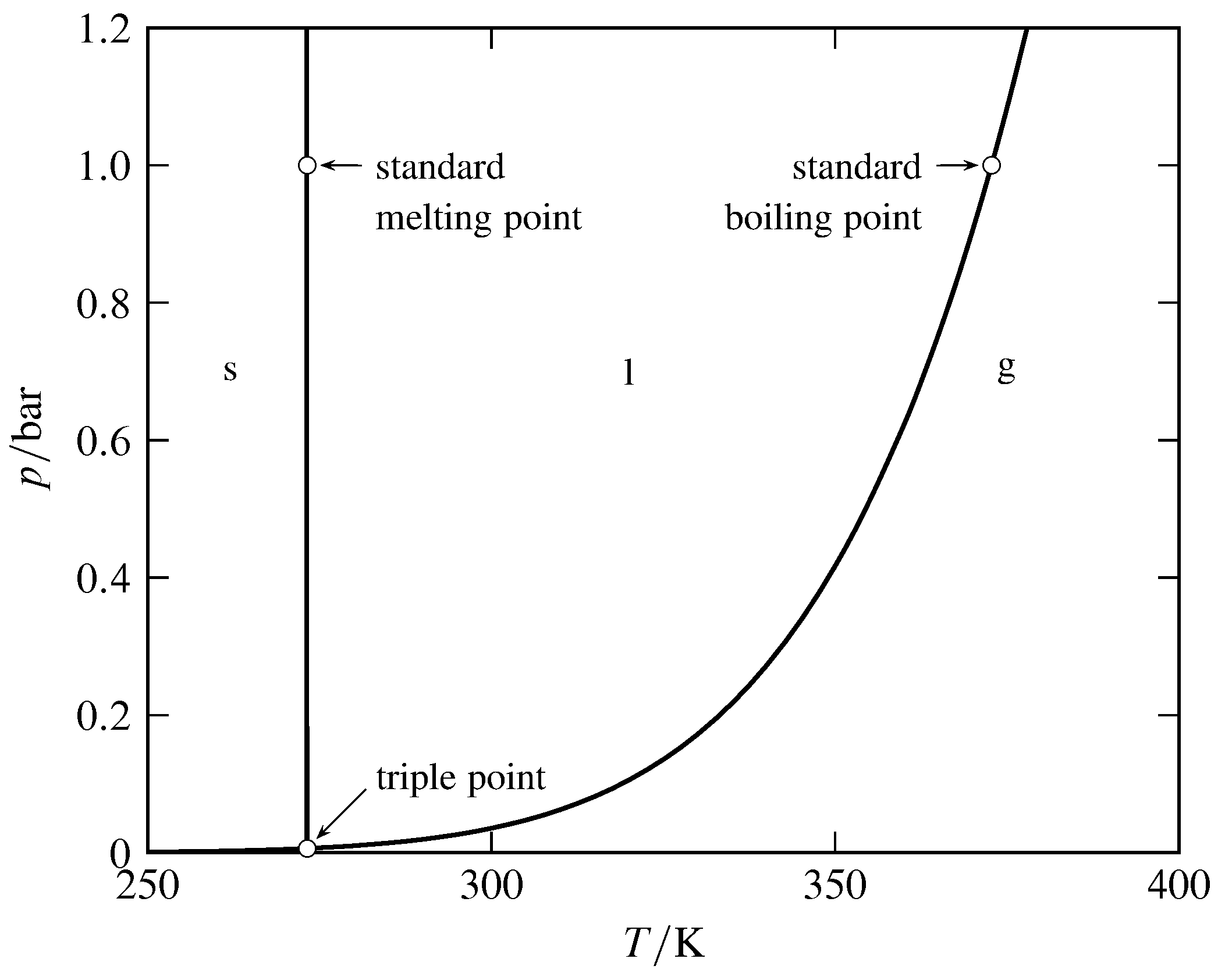
Figura 8.6 Diagrama de fases presión-temperatura de H\(_2\) O. (Basado en datos en NIST Chemistry WebBook.)
La relación entre temperatura y presión en un sistema con dos fases en equilibrio se muestra mediante la curva de coexistencia que separa las dos áreas monofásicas en el diagrama presión-temperatura (ver Fig. 8.6). Considere la curva líquido-gas. Si pensamos en la variable independiente, la curva es una curva vapor-presión que muestra cómo la presión de vapor del líquido varía con la temperatura.\(T\) Si, sin embargo,\(p\) es la variable independiente, entonces la curva es una curva de punto de ebullición que muestra la dependencia del punto de ebullición de la presión.
El punto de fusión o punto de ebullición normal se refiere a una presión de una atmósfera, y el punto de fusión estándar o punto de ebullición se refiere a la presión estándar. Así, el punto de ebullición normal del agua (\(99.97\units{\(\degC\)}\)) es el punto de ebullición en\(1\units{atm}\); esta temperatura también se conoce como el punto de vapor. El punto de ebullición estándar del agua (\(99.61\units{\(\degC\)}\)) es el punto de ebullición a la presión ligeramente más baja de\(1\br\).
Las curvas de convivencia se discutirán más a fondo en la Sec. 8.4.
8.2.3 El punto crítico
Cada sustancia tiene una cierta temperatura, la temperatura crítica, por encima de la cual solo puede existir una fase fluida a cualquier volumen y presión (Sec. 2.2.3). El punto crítico es el punto en un diagrama de fases correspondiente a la coexistencia líquido-gas a la temperatura crítica, y la presión crítica es la presión en este punto.

Figura 8.7 Bulbo de vidrio lleno de CO\(_2\) a un valor\(V/n\) cercano al valor crítico, visto a cuatro temperaturas diferentes. Las tres bolas tienen densidades menores, aproximadamente iguales y mayores que la densidad crítica. (Fotos con permiso del fotógrafo; aparecieron en Jan V. Sengers y Anneke Levelt Sengers, Chem. Ing. Noticias, 10 de junio de 104—118, 1968.)
(a) Fluido supercrítico a una temperatura superior a la temperatura crítica.
b) Opalescencia intensa justo por encima de la temperatura crítica.
c) Formación de menisco ligeramente por debajo de la temperatura crítica; líquido y gas de casi la misma densidad.
d) Temperatura muy por debajo de la temperatura crítica; líquidos y gases de densidades muy diferentes.
Para observar experimentalmente el punto crítico de una sustancia, podemos evacuar un recipiente de vidrio, introducir una cantidad de la sustancia tal que\(V/n\) sea aproximadamente igual al volumen molar en el punto crítico, sellar el recipiente y elevar la temperatura por encima de la temperatura crítica. El recipiente ahora contiene una sola fase fluida. Cuando la sustancia se enfría lentamente a una temperatura ligeramente superior a la temperatura crítica, presenta un aspecto turbio, fenómeno llamado opalescencia crítica (Fig. 8.7). La opalescencia es la dispersión de la luz causada por grandes fluctuaciones de densidad local. A la temperatura crítica, se forma un menisco entre las fases líquida y gaseosa de prácticamente la misma densidad. Con un enfriamiento adicional, la densidad del líquido aumenta y la densidad del gas disminuye.
A temperaturas por encima de la temperatura crítica y presiones por encima de la presión crítica, la fase fluida existente se denomina fluido supercrítico. Así, un fluido supercrítico de una sustancia pura es un fluido que no experimenta una transición de fase a una fase fluida diferente cuando cambiamos la presión a temperatura constante o cambiamos la temperatura a presión constante.
Sin embargo, si aumentamos\(p\) a constante\(T\), el fluido supercrítico cambiará a un sólido. En el diagrama de fases de H\(_2\) O, la curva de coexistencia para hielo VII y líquido que se muestra en la Fig. 8.4 se extiende a una temperatura superior a la temperatura crítica de\(647\K\). Así, el agua supercrítica se puede convertir en hielo VII por compresión isotérmica.
Un fluido en la región supercrítica puede tener una densidad comparable a la del líquido, y puede ser más compresible que el líquido. En condiciones supercríticas, una sustancia suele ser un excelente solvente para sólidos y líquidos. Al variar la presión o temperatura, se puede cambiar el poder de solvatación; al reducir la presión isotérmicamente, la sustancia se puede eliminar fácilmente como un gas de los solutos disueltos. Estas propiedades hacen que los fluidos supercríticos sean útiles para la cromatografía y extracción con disolventes.
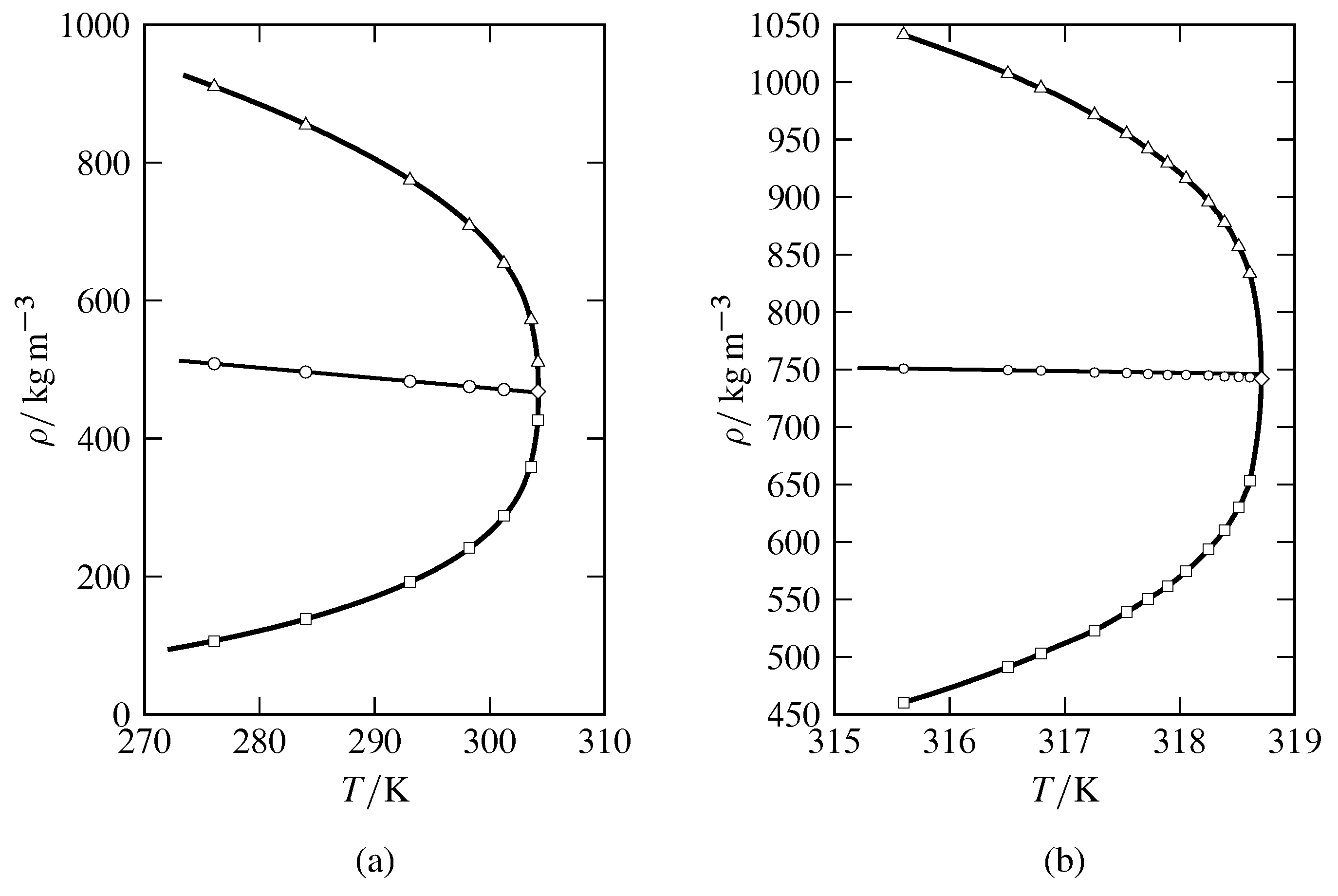
Figura 8.8 Densidades de fases gaseosas y líquidas coexistentes cercanas al punto crítico como funciones de temperatura para (a) CO\(_2\) (basado en datos de A. Michels, B. Blaisse y C. Michels, Proc. R. Soc. Londres, Ser. A, 160, 358—375, 1937); (b) SF\(_6\) (datos de M. W. Pestak et al, Phys. Rev. B, 36, 599—614, 1987, Cuadro VII). Las densidades de gas experimentales se muestran por cuadrados abiertos y las densidades de líquidos experimentales por triángulos abiertos. La densidad media a cada temperatura experimental se muestra mediante un círculo abierto. El diamante abierto está a la temperatura crítica y densidad crítica.
La temperatura crítica de una sustancia se puede medir con bastante precisión observando la aparición o desaparición de un menisco líquido-gas, y la presión crítica se puede medir a esta temperatura con un manómetro de alta presión. Para evaluar la densidad en el punto crítico, lo mejor es extrapolar la densidad media de las fases líquida y gaseosa coexistentes\((\rho\sups{l} +\rho\sups{g})/2\), a la temperatura crítica como se ilustra en la Fig. 8.8. La observación de que la densidad media se aproxima estrechamente a una función lineal de la temperatura, como se muestra en la figura, se conoce como la ley de los diámetros rectilíneos, o la ley de Cailletet y Matthias. Esta ley es una aproximación, como puede verse por la pequeña desviación de la densidad media de SF\(_6\) de una relación lineal muy cercana al punto crítico en la Fig. 8.8 (b). Este fracaso de la ley de diámetros rectilíneos es pronosticado por tratamientos teóricos recientes (Jingtao Wang y Mikhail A. Anisimov, Phys. Rev. E, 75, 051107, 2007; Hassan Behnejad, Jan V. Sgers, y Mikhail A. Anisimov, en A. R. H. Goodwin, J. V. Sgers, y C. J. Peters, editores, Termodinámica Aplicada de Fluidos, páginas 321—367, Royal Society of Chemistry, Cambridge, 2010).
8.2.4 La regla de la palanca
Considere un sistema de una sola sustancia cuyo punto del sistema esté en un área bifásica de un diagrama de fases presión-volumen. ¿Cómo podemos determinar los montos en las dos fases?
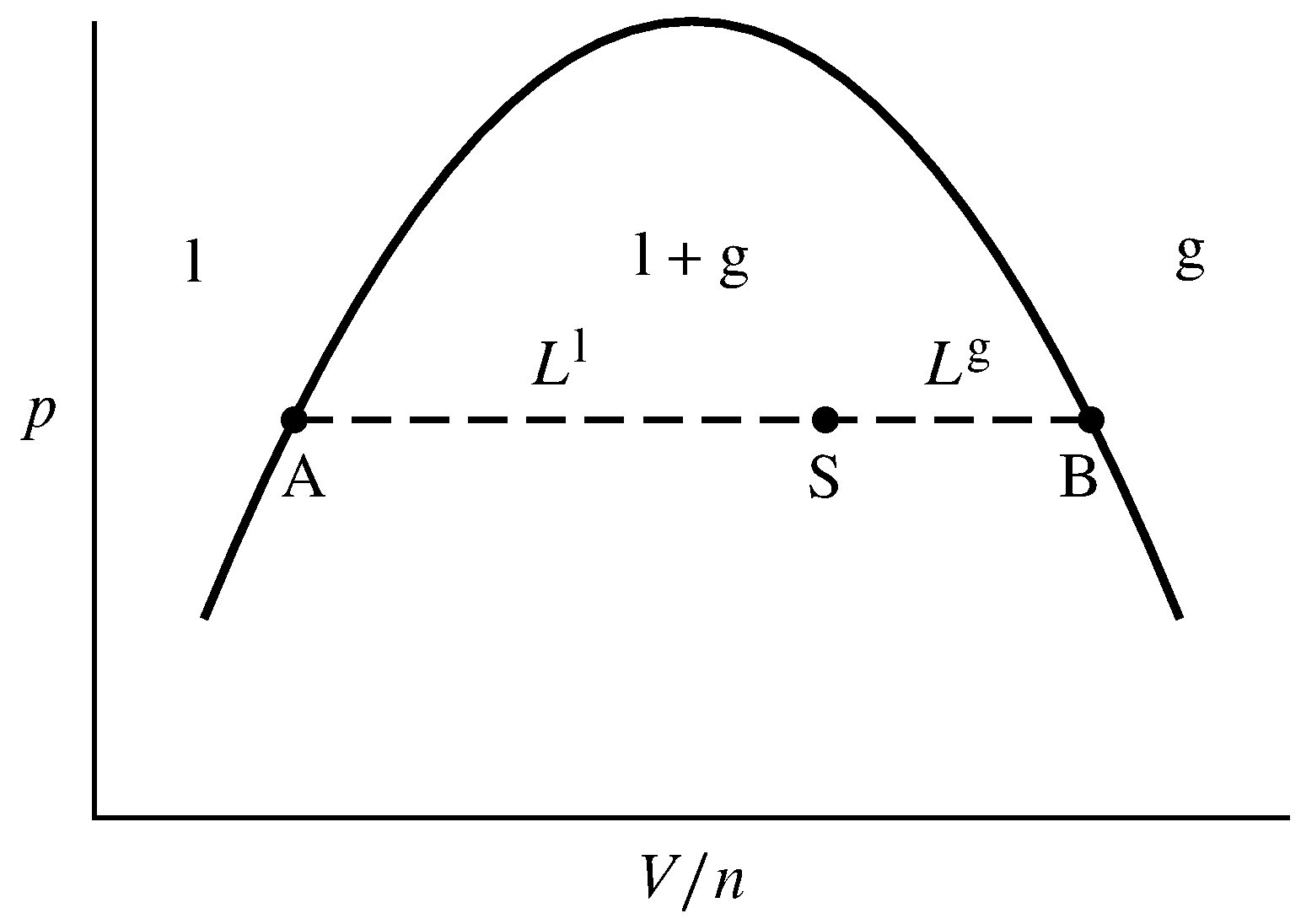
Figura 8.9 Línea de unión (discontinua) en constante\(T\) y\(p\) en el área líquido-gas de un diagrama de fases presión-volumen. Los puntos A y B están en los extremos de la línea de empate, y el punto S es un punto del sistema en la línea de empate. \(L\sups{l}\)y\(L\sups{g}\) son las longitudes AS y SB, respectivamente.
Como ejemplo, dejar que el sistema contenga una cantidad fija\(n\) de una sustancia pura dividida en fases líquida y gaseosa, a una temperatura y presión a las que estas fases puedan coexistir en equilibrio. Cuando el calor se transfiere al sistema en este\(T\) y\(p\), parte del líquido se vaporiza por una transición de fase líquido-gas y\(V\) aumenta; la extracción de calor en esto\(T\) y\(p\) hace que el gas se condense y\(V\) disminuya. Los volúmenes molares y otras propiedades intensivas de las fases líquida y gaseosa individuales permanecen constantes durante estos cambios a constante\(T\) y\(p\). En el diagrama de fases presión-volumen de la Fig. 8.9, los cambios de volumen corresponden al movimiento del punto del sistema hacia la derecha o hacia la izquierda a lo largo de la línea de unión AB.
Cuando se transfiere suficiente calor al sistema para vaporizar todo el líquido en el momento dado\(T\) y\(p\), el punto del sistema se mueve al punto B en el extremo derecho de la línea de unión. \(V/n\)en este punto debe ser el mismo que el volumen molar del gas,\(V\m\sups{g}\). Podemos ver esto porque el punto del sistema podría haberse movido desde dentro del área de gas monofásico a esta posición en el límite sin sufrir una transición de fase.
Cuando, por otro lado, se transfiere suficiente calor fuera del sistema para condensar todo el gas, el punto del sistema se mueve al punto A en el extremo izquierdo de la línea de unión. \(V/n\)en este punto es el volumen molar del líquido,\(V\m\sups{l}\).
Cuando el punto del sistema está en la posición S en la línea de unión, tanto el líquido como el gas están presentes. Sus cantidades deben ser tales que el volumen total sea la suma de los volúmenes de las fases individuales, y la cantidad total sea la suma de las cantidades en las dos fases:\ begin {ecuación} V = V\ sups {l} +V\ sups {g} = n\ sups {l} V\ m\ sups {l} +n\ sups {g} V\ m\ sups {g} tag\ {8.2.1}\ final {ecuación}\ comenzar {ecuación} n = n\ sups {l} +n\ sups {g}\ tag {8.2.2}\ end {ecuación} El valor de\(V/n\) en el punto del sistema viene dado entonces por la ecuación\ begin {ecuación}\ frac {V} {n} =\ frac {n\ sups {l} V\ m\ sups {l} +n\ sups {g} V\ m\ sups {g}} {n\ sups {l} +n\ sups ps {g}}\ tag {8.2.3}\ end {ecuación} que se puede reorganizar para\ comenzar {ecuación} n\ sups {l}\ left (V\ m\ sups {l} -\ frac {V} {n}\ derecha) =n\ sups {g}\ izquierda (\ frac {V} {n} -V\ m\ sups {g}\ derecha)\ tag {8.2.4}\ end {ecuación} Las cantidades\(V\m\sups{l}-V/n\) y\(V/n-V\m\sups{g}\) son las longitudes\(L\sups{l}\) y\(L\sups{g}\), respectivamente, definidas en la figura y medidas en unidades de\(V/n\). Esto nos da la regla de palanca para el equilibrio líquido-gas:\ begin {recoger}\ s {n\ sups {l} L\ sups {l} = n\ sups {g} L\ sups {g}\ cuádruple\ tx {o}\ quad\ frac {n\ sups {g}} {n\ sups {l}} =\ frac {L\ sups {l}} L\ sups {g}}}\ tag {8.2.5}\ cond {(coexistiendo líquido y gas}\ nextcond {fases de una sustancia pura)}\ end {reúnen} ( La relación se llama regla de palanca por analogía con una palanca mecánica estacionaria, cada extremo de la cual tiene el mismo valor del producto de la fuerza aplicada y distancia desde el fulcro.)
En la Fig. 8.9 el punto S del sistema se posiciona en la línea de empate dos tercios del camino desde el extremo izquierdo, haciendo que la longitud sea el\(L\sups{l}\) doble de larga\(L\sups{g}\). La regla de palanca da entonces la relación de montos:\(n\sups{g}/n\sups{l} = L\sups{l}/L\sups{g} =2\). Un tercio de la cantidad total es líquida y dos tercios es gas.
No podemos aplicar la regla de palanca a un punto en la línea triple, porque necesitamos más que el valor de\(V/n\) para determinar los montos relativos presentes en tres fases.
Podemos derivar una forma más general de la regla de palanca que se necesitará en el capítulo 13 para diagramas de fase de sistemas multicomponentes. Esta forma general se puede aplicar a cualquier área bifásica de un diagrama de fases bidimensional en el que sea válida una construcción de línea de enlace, con la posición del punto del sistema a lo largo de la línea de empate dada por la variable\ begin {ecuación} F\ defn\ frac {a} {b}\ tag {8.2.6}\ end {ecuación} donde\(a\) y\(b\) son extensas funciones estatales. (En el diagrama de fases presión-volumen de la Fig. 8.9, estas funciones son\(a=V\) y\(b=n\) y la posición del punto del sistema viene dada por\(F=V/n\).) Repetimos los pasos de la derivación anterior, etiquetando las dos fases por superíndices\(\pha\) y\(\phb\) en lugar de\(l\) y\(g\). La relación correspondiente a la Ecuación 8.2.4 es\ begin {ecuación} b\ aph (F\ aph-F) =b\ bph (F-F\ bph)\ tag {8.2.7}\ end {ecuación} Si\(L\aph\) y\(L\bph\) son longitudes medidas a lo largo de la línea de enlace desde el punto del sistema hasta los extremos de la línea de enlace en fases simples\(\pha\) y\(\phb\), respectivamente, la Ec. 8.2.7 es equivalente a la regla general de palanca\ begin {ecuación} b\ aph L\ aph = b\ bph L\ bph\ qquad\ tx {o}\ qquad\ frac {b\ bph} {b\ aph} =\ frac {L\ aph} {L\ bph}\ tag {8.2.8}\ end {ecuación}
8.2.5 Propiedades de volumen
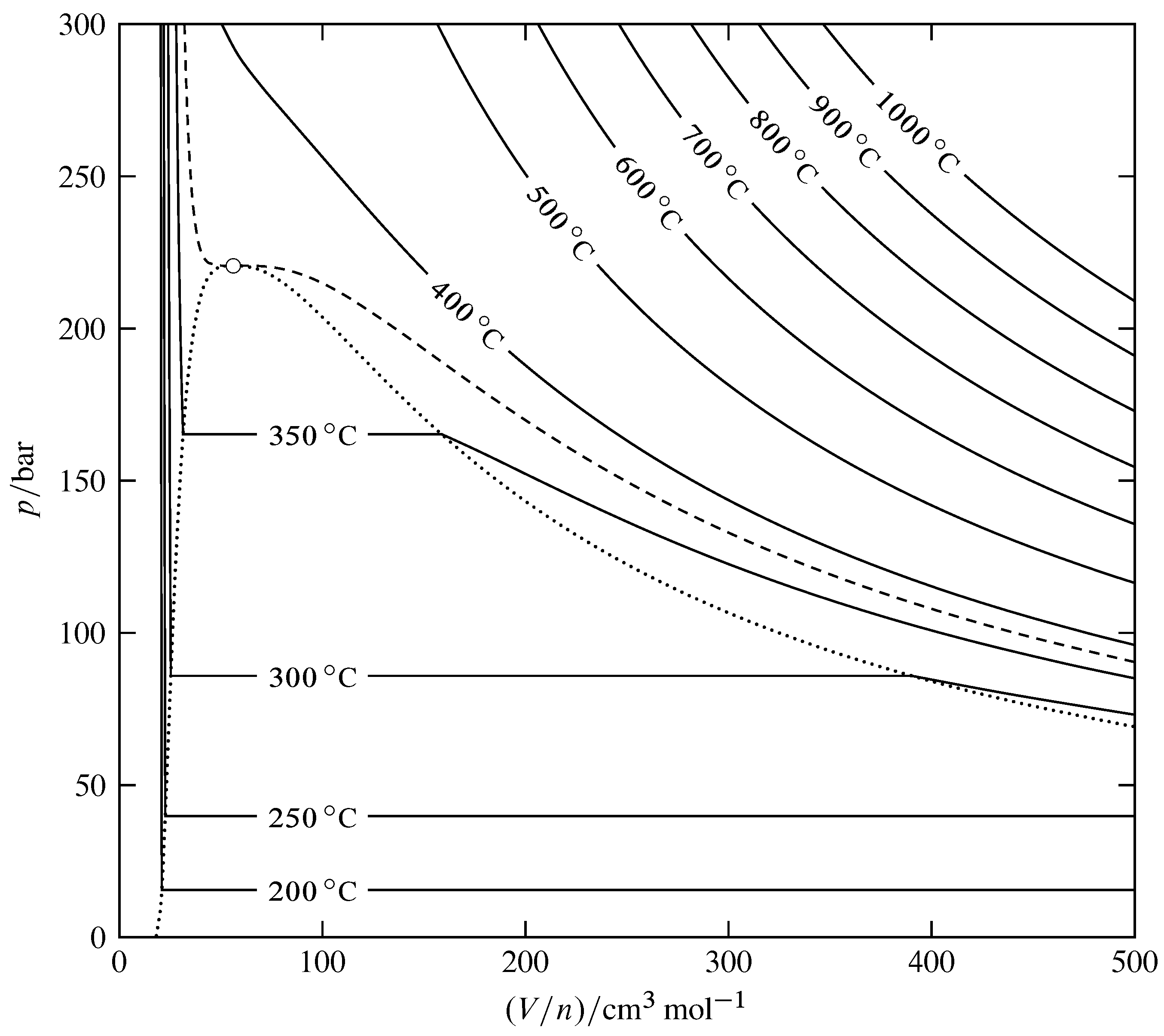
Figura 8.10 Isotermas para las fases fluidas de H\(_2\) O (basado en datos en NIST Chemistry WebBook). El círculo abierto indica el punto crítico, la curva discontinua es la isoterma crítica en\(373.95\units{\(\degC\)}\), y la curva punteada encierra el área bifásica del diagrama de fases presión-volumen. La triple línea se encuentra demasiado cerca de la parte inferior del diagrama para ser visible en esta escala.
La Figura 8.10 es un diagrama de fases presión-volumen para H\(_2\) O. En el diagrama se dibujan isotermas (curvas de constante\(T\)). Estas isotermas definen la forma de la\(T\) superficie tridimensional\(p\).\((V/n)\) El área que contiene los segmentos de isoterma horizontal es el área bifásica para las fases líquida y gaseosa coexistentes. El límite de esta área se define por la curva punteada dibujada a través de los extremos de los segmentos horizontales. El área líquida monofásica se encuentra a la izquierda de esta curva, el área de gas monofásico se encuentra a la derecha y el punto crítico se encuentra en la parte superior.
El diagrama contiene la información necesaria para evaluar el volumen molar a cualquier temperatura y presión en la región monofásica y las derivadas del volumen molar con respecto a temperatura y presión. En un punto del sistema en la región monofásica, la pendiente de la isoterma que pasa por el punto es la derivada parcial\(\pd{p}{V\m}{T}\). Dado que la compresibilidad isotérmica viene dada por\(\kT = -(1/V\m)\pd{V\m}{p}{T}\), tenemos\ begin {ecuación}\ kT = -\ frac {1} {V\ m\ times\ tx {pendiente de isoterma}}\ tag {8.2.9}\ end {ecuación} Vemos en la Fig. 8.10 que las pendientes de las isotermas son grandes y negativas en la región líquida, menores y negativas en el gas y supercríticas regiones fluidas, y acercarse a cero en el punto crítico. En consecuencia, la compresibilidad isotérmica del gas y del fluido supercrítico es mucho mayor que la del líquido, acercándose al infinito en el punto crítico. La opalescencia crítica observada en la Fig. 8.7 es causada por fluctuaciones de densidad local, las cuales son grandes cuando\(\kT\) son grandes.
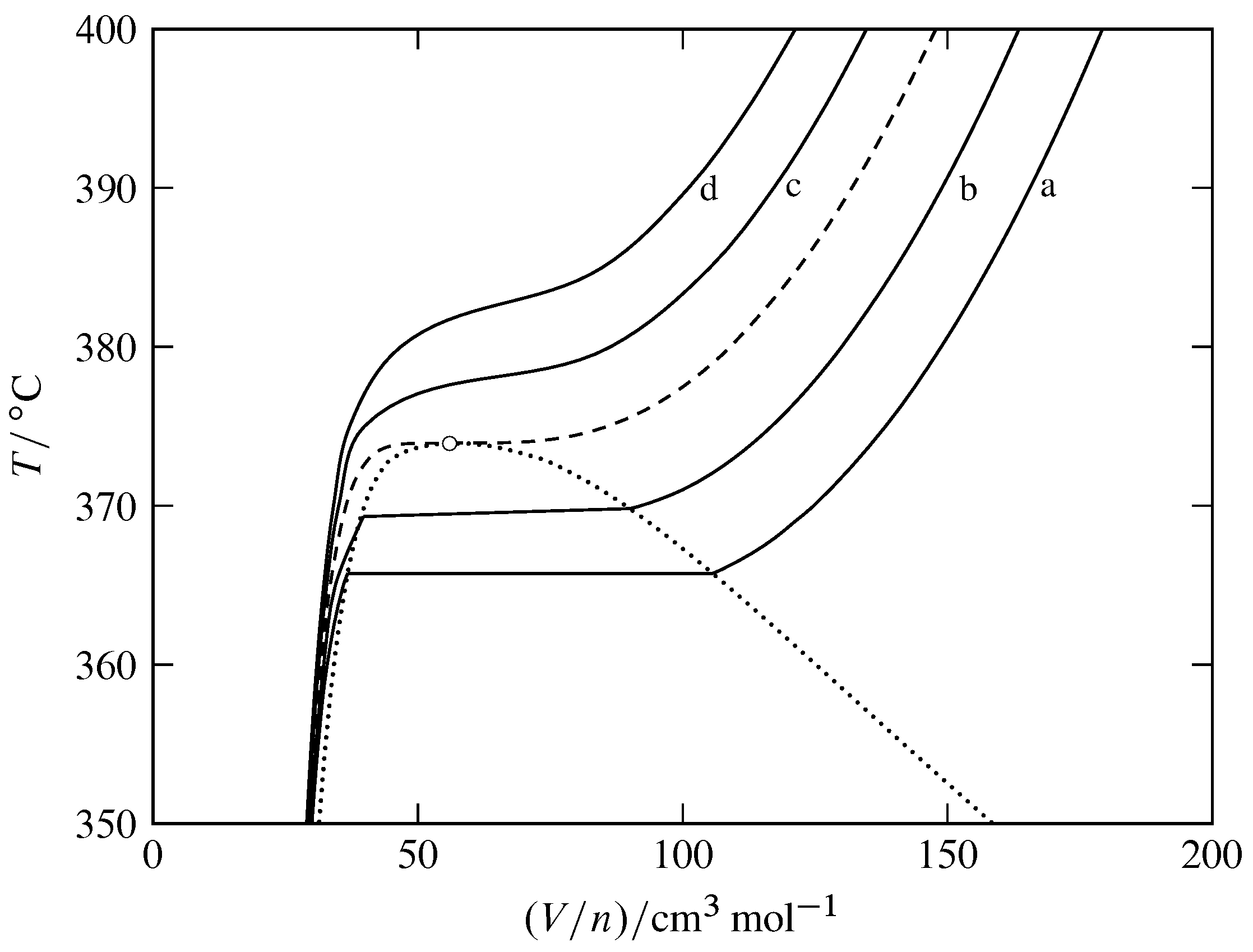
Figura 8.11 Isobares para las fases fluidas de H\(_2\) O (basado en datos en NIST Chemistry WebBook). El círculo abierto indica el punto crítico, la curva discontinua es la isóbar crítica en\(220.64\br\), y la curva punteada encierra el área bifásica del diagrama de fases temperatura-volumen.
Curvas sólidas: a,\(p=200\br\); b,\(p=210\br\); c,\(p=230\br\); d,\(p=240\br\).
La Figura 8.11 muestra isobarras para H\(_2\) O en lugar de isotermas. En un punto del sistema en la región monofásica, la pendiente de la isobarra que pasa por el punto es la derivada parcial\(\pd{T}{V\m}{p}\). El coeficiente de expansión cúbica\(\alpha\) es igual a\((1/V\m)\pd{V\m}{T}{p}\), así tenemos\ begin {ecuación}\ alpha =\ frac {1} {V\ m\ times\ tx {pendiente de isobar}}\ tag {8.2.10}\ end {ecuación} La figura muestra que las pendientes de las isobarras son grandes y positivas en la región líquida, menores y negativas en el gas y regiones de fluido supercrítico, y acercarse a cero en el punto crítico. Así, el gas y el fluido supercrítico tienen coeficientes de expansión cúbica mucho mayores que el líquido. El valor de\(\alpha\) se aproxima al infinito en el punto crítico, lo que significa que en la región crítica la distribución de la densidad se ve muy afectada por los gradientes de temperatura. Esto puede explicar la posición baja de la bola media en la Fig. 8.7 (b).