
4: La lógica de la síntesis




- Última actualización
- 30 oct 2022
- Guardar como PDF
( \newcommand{\kernel}{\mathrm{null}\,}\)
'Podría haber ARTE en la síntesis orgánica' declaró el inimitable monarca de la síntesis orgánica, el profesor R.B. Woodward. Su escuela dio a conocer varios enfoques elegantes que cubren una variedad de estructuras complejas y abrió nuevos terrenos para definir el arte de la síntesis orgánica. 'Si la síntesis orgánica es una rama de la ciencia, ¿cuál es la LÓGICA de la síntesis orgánica? ' maravilló a varios otros. El desarrollo del concepto de enfoques lógicos hacia la síntesis ha ido evolucionando en las últimas décadas. Algunos incondicionales centraron su atención en este tema e intentaron evolucionar un patrón para definir esta lógica. No cabe duda de que todos los que incursionamos con la síntesis aportamos nuestro pedacito en la magnífica dirección. Algunos nombres destacan en nuestras mentes por sus destacadas contribuciones. Las contribuciones notables provinieron de las escuelas de J.A. Mariscal, E.J. Wenkert, G. Stock, S Hanessian, E.E. van Tamalen, S. Masamune, R.B. Woodward, E.J. Corey y varios otros. Más enfocados en este tema fueron las contribuciones de la escuela de E.J. Corey.
El periodo 1960 — 1990 fue testigo de la evolución de este pensamiento y el concepto floreció hasta convertirse en un tema de pleno derecho que ahora merece un espacio separado en el currículo universitario. Los desarrollos anteriores se centraron en la idea de ENFOQUES ANTITÉTICOS y perfeccionaron el arte de la DESCONEXIÓN vía Esto condujo a enfoques lógicos para la construcción de ÁRBOLES SINTÉTICOS que resumieron diversos enfoques posibles para la estructura de Objetivo propuesta. Todas las desconexiones pueden no conducir a buenas rutas de síntesis. Una vez construido el árbol sintético, se analizaron críticamente las ramas individuales. Se investigaron las reacciones involucradas, para estudiar su factibilidad en el laboratorio, se analizaron sus vías mecanicistas para comprender las implicaciones conformacionales y estereoquímicas sobre el resultado de cada paso involucrado y también se estimaron los factores tiempo/costo de las rutas propuestas. Se identificaron las posibles áreas de escollo y se escaneó críticamente la literatura para asegurarse de que los pasos contemplados ya eran conocidos o factibles sobre la base de la química conocida. En algunos casos, los compuestos modelo se construyeron primero para estudiar la viabilidad de la reacción particular, antes de embarcarse en la síntesis de la compleja arquitectura molecular. Por lo tanto, ahora se pone en marcha un largo proceso de planeación lógica antes del inicio del proyecto sintético real. A pesar de todas estas cuidadosas y largas preparaciones, un químico experimentado todavía está cansado de la Espada de síntesis de Damocles, a saber, el probable fracaso de un paso crítico en la ruta (s) propuesta (s), resultando en un fracaso total de todo el proyecto. Todos los logros son 10% inspiración y 90% transpiración. Para estos valientes ingenieros moleculares, a veces también llamados químicos, estos programas de larga data y los posibles peligros de fallas aún valen, porque la transpiración es suficiente recompensa.
Un conocimiento sólido de la química orgánica mecanicista, información detallada sobre el arte y la ciencia de las transformaciones de grupos funcionales, la formación de enlaces y las reacciones de escisión, el dominio sobre las técnicas de separación y purificación y un conocimiento sólido del análisis espectroscópico son conceptos básicos esenciales para la síntesis de moléculas. Un químico sintético también debe estar al tanto de los desarrollos en estrategias sintéticas generadas a lo largo de los años para diferentes grupos de compuestos, que incluyen Reglas y lineamientos que rigen la síntesis. Dado que la química orgánica tiene un fuerte impacto en el desarrollo de otras disciplinas hermanas como la farmacia, la bioquímica y la ciencia de los materiales, la capacidad de comprender una o más de estas áreas e interactuar con ellas usando sus terminologías también es una virtud añadida para un químico sintético. Con logros desde la síntesis de moléculas tensas (antes consideradas difíciles (si no imposibles) de sintetizar, hasta la síntesis de moléculas complejas, altamente funcionalizadas e inestables, un químico orgánico podría ahora decir con confianza que podría sintetizar cualquier molécula que sea teóricamente factible. Este es el estado actual del poder de la síntesis orgánica. Con base en la tarea asignada al químico, seleccionaría una molécula Target para su investigación e idearía vías adecuadas para la síntesis.
Estrategias de Protección y Desprotección en Síntesis Orgánica
Para la manipulación de grupos funcionales y formación de nuevos enlaces covalentes se hace uso de un gran número de Reactivos y Reacciones de Nombre. En síntesis orgánicas complejas, los materiales de partida y los intermedios en el esquema sintético suelen tener más de un grupo funcional reactivo. A continuación se muestran algunos de estos bloques de construcción multifuncionales para ilustrar este punto (Fig. 4.1.1). Mientras se trabaja en tan complejo
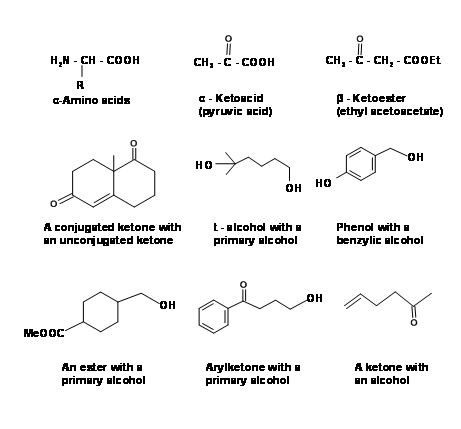
moléculas, a menudo es necesario proteger algunos grupos para permitir el trabajo selectivo solo en las ubicaciones deseadas. Los químicos orgánicos han confiado en gran medida en tales estrategias de protección/desprotección y han desarrollado diligentemente protocolos de protección (enmascaramiento) y desprotección (desenmascaramiento). Discutiremos algunos de los grupos protectores importantes en este capítulo.
Antes de continuar, hay que recalcar aquí que este protocolo debe aplicarse sólo después de que se hayan analizado críticamente las opciones alternas. Esto se debe a que la estrategia de protección/desprotección implica un aumento de al menos dos pasos críticos más, lo que se suma a la duración de la síntesis y la consiguiente caída en los rendimientos globales del compuesto deseado. En reacciones a gran escala, esto lleva a un enorme impacto en la Economía Atom y al costo de contaminación del proceso sintético. Todo esto se traduce en un incremento en el costo general de la molécula de fármaco final.
Estrategias de Protección
Reactivo Selectivo de Grupo/Sitio: No siempre se requiere protección/desprotección siempre que vea una multiplicidad de grupos funcionales. Se podría resolver el problema de selectividad mediante el uso de reacciones selectivas de sitio/reactivos. Al elegir un reactivo selectivo apropiado que se adapte al esquema en cuestión, podría atacar selectivamente solo uno de los sitios reactivos. Considera una cetona olefínica (Fig 4.1.2). La reducción de borohidruro de sodio en metanol como disolvente podría reducir selectivamente el grupo ceto-a un alcohol secundario
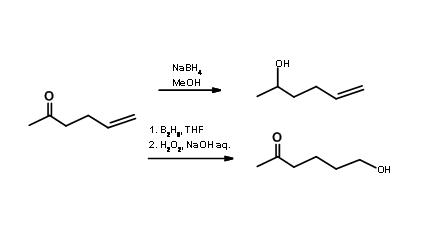
dejando la olefina intacta. Por otro lado, el reactivo de diborano en THF como disolvente sería un reactivo de elección cuando se desea la reducción selectiva en el resto olefínico. La reducción de diborano de una olefina es varias veces más rápida que la reducción de cetonas. La escisión oxidativa del producto borano también es selectiva. De esta manera, se puede evitar la estrategia de protección/desprotección empleando un reactivo selectivo. En las reacciones de formación de enlaces C — C nos encontramos con varios reactivos selectivos de sitio de este tipo. Uno de esos reactivos ampliamente utilizado en la investigación es el reactivo de Wittig. Atacan el aldehído o cetona selectivamente en presencia de éster, nitrilo. olefina etc.
Protección Selectiva
En el caso de una molécula como 4.1.3A (Fig 4.1.3) que porta una olefina y un ácido carboxílico, el grupo -COOH es varias veces más reactivo que la olefina hacia la reducción de diborano.
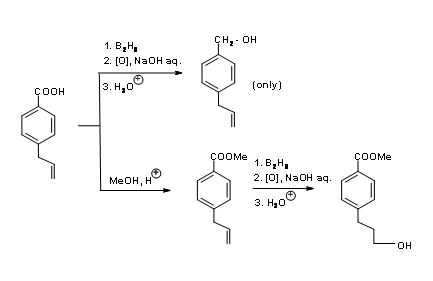
La hidroboración/oxidación reduce el ácido a un alcohol primario, dejando la olefina intacta. Por otro lado, si se necesita una reducción selectiva de olefina, el grupo ácido tiene que procesarse a través de una secuencia selectiva de protección/desprotección como se muestra en (Fig 4.1.3)
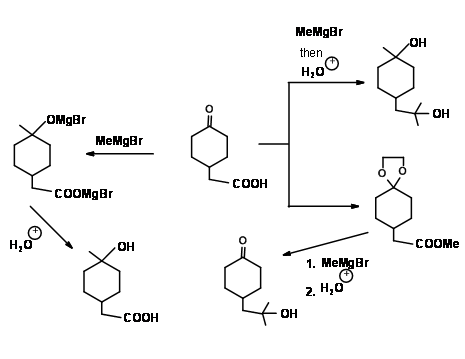
El Compuesto 4.1.4A ilustra varios puntos importantes en el protocolo de Protección/Desprotección. Ambos grupos funcionales podrían reaccionar con un Reactivo de Grignard. El grupo ácido carboxílico reaccionaría primero con un mol del reactivo de Grignard para dar una sal de anión carboxilato. Este anión no reacciona más con el reactivo. Cuando se añaden dos moles de reactivo de Grignard a la mezcla de reacción, el segundo mol ataca a la cetona para dar un alcohol terciario. En el tratamiento acuoso, el grupo ácido se regenera. Así, el primer mol del reactivo proporciona una protección transitoria selectiva para el grupo -COOH. Una vez esterificado el grupo ácido, se pierde dicha selectividad hacia este reactivo. El reactivo ataca en ambos sitios. Si se desea la reacción solo en el sitio éster, el grupo ceto-debe protegerse selectivamente como un acetal. En el siguiente paso, se lleva a cabo la reacción de grignard. Ahora el reactivo solo tiene un grupo disponible para la reacción. En el tratamiento con ácido, la protección cetal en el compuesto intermedio también se hidroliza para regenerar el grupo cetal.
Protección Ortogonal o Protección Diferencial
La protección ortogonal es una estrategia que permite la desprotección de múltiples grupos protectores uno a la vez, cada uno con un conjunto dedicado de reactivos y/condiciones de reacción sin afectar al otro. Esta técnica se ilustra mejor con la formación de enlaces peptídicos y reacciones de desprotección asociadas. Un aminoácido tiene dos grupos funcionales —N H 2 y —COOH. Cuando dos aminoácidos (A y B) reaccionan en condiciones para la reacción de condensación del enlace peptídico, se podría formar una mezcla de 4 dipéptidos (al menos) como se muestra a continuación.
A+B→A−A+A−B+B−A+B−B
Si nos interesa un solo producto A — B, tenemos que hacer protecciones selectivas y desprotecciones selectivas en una secuencia adecuada. Considere la siguiente reacción de formación de enlaces peptídicos.
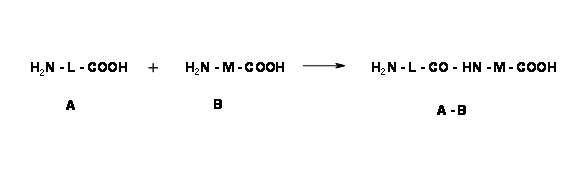
Para obtener solo un producto A — B, debemos proteger el N — terminal de 'A' y C — terminal de 'B'. Veamos de cerca dos esquemas de formación de dipéptidos diferentes. En la siguiente secuencia, el C-terminal está protegido de dos maneras diferentes para un aminoácido. Para el segundo aminoácido, el N-terminal está protegido con una protección Boc- lábil a los ácidos.
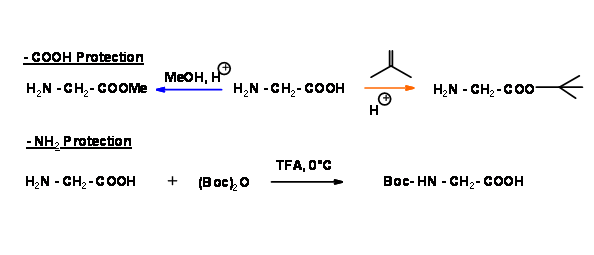
En la siguiente etapa, los dos aminoácidos monoprotegidos se acoplan como se muestra a continuación.
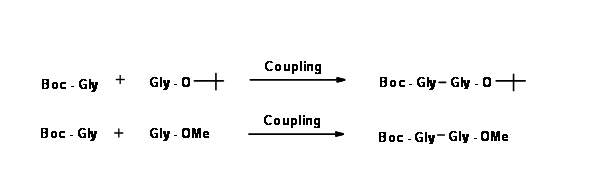
Echa un vistazo de cerca a ambos productos. En el primer producto, ambas protecciones son sensibles a los ácidos. Si el producto final deseado es el dipéptido libre de protección, esta es de hecho una ruta corta.
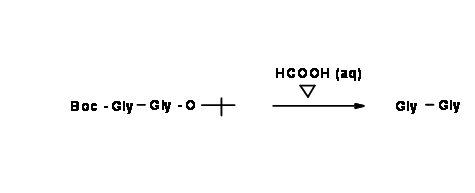
Si el producto deseado es un dipéptido monoprotegido, entonces la desprotección selectiva es la reacción preferida. Esto es factible solo cuando utilizamos compuestos de partida que están protegidos diferencialmente. Esto se llama Protección Ortogonal.
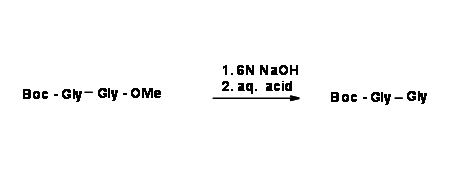
Técnicas similares también están disponibles para otros grupos funcionales. Ahora aprendamos más sobre Protección/Desprotección para algunos grupos funcionales importantes.
Protección de R — Grupo COOH
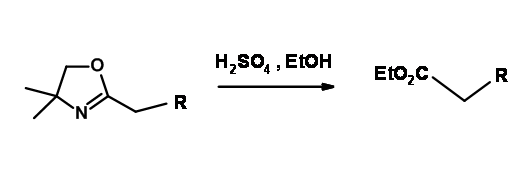
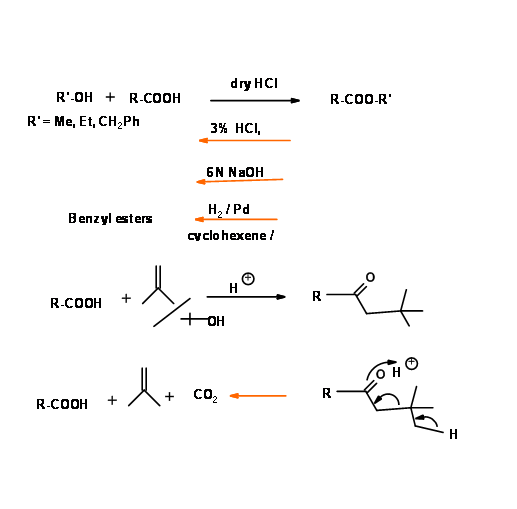
En la introducción, hemos visto que el ion carboxilato brinda protección a un ataque de reactivos de Grignard en este carbono carbonilo. Sin embargo, esto no es suficiente para una gran variedad de reactivos. Las 2-oxazolinas de Meyer enmascaran una función ácida mientras activan la posición α para la reacción de litiación. El uso de este grupo como protección para el grupo —COOH es raro.
Protección de Aldehídos y Cetonas
Dado que los alcoholes, aldehídos y cetonas son los grupos funcionales más frecuentemente manipulados en la síntesis orgánica, ha aparecido mucho trabajo en sus estrategias de protección/desprotección. En esta discusión vamos a enfocarnos en las clases de grupos protectores más que en un tratamiento exhaustivo de todas las protecciones.
Acetales
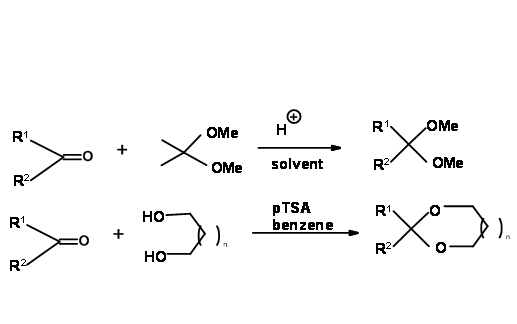
Existen dos métodos generales para la introducción de esta protección. La transcetalación es el método de elección cuando se desean acetales (cetales) con metanol. La acetona es el subproducto, el cual tiene que ser eliminado para desplazar el equilibrio hacia el lado derecho. Esto se logra calentando a reflujo con un gran exceso del reactivo acetónido. La acetona formada se destila constantemente. En el caso de los dioles cíclicos, el agua formada se elimina continuamente utilizando un condensador Dean-Stork (Fig 4.1.6).
La tasa de formación de cetales a partir de cetonas y 1,2-etandiol (etilenglicol), 1,3-propanodiol y 2,2-dimetil-1,3-propanodiol son diferentes. Así es la reacción de decetalación. Esto ha permitido que los químicos trabajen selectivamente en un solo centro. Los siguientes ejemplos de la química de esteroides ilustran estos puntos (Fig. 4.1.7).
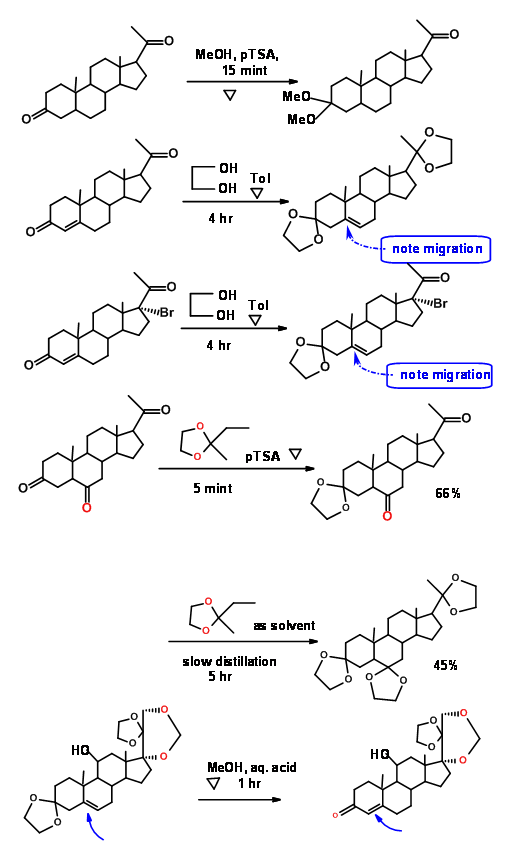
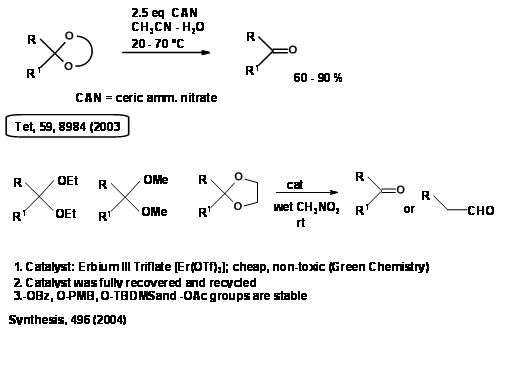
La demanda de procesos de Química Verde ha impulsado la búsqueda de nuevos procedimientos verdes. Aquí se dan algunos ejemplos de literatura reciente (Fig. 4.1.8).
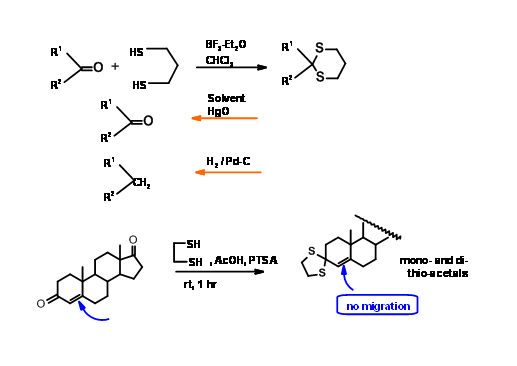
Tiocetales
En comparación con sus análogos de oxígeno, los tiocetales difieren notablemente en su química. La formación así como la desprotección son promovidas por ácidos de Lewis adecuados. Los tioacetales son marcadamente estables bajo condiciones de decetalación, allanando así el camino para operaciones selectivas en dos centros diferentes. Cuando las cetonas conjugadas están involucradas, la formación de cetales (así como la desprotección) procede con migración de doble enlace. Por otro lado, los tiocetales se forman y decetalan sin migración de doble enlace (Fig 4.1.9).
Protección de grupos Amino (-NH2 y -NH)
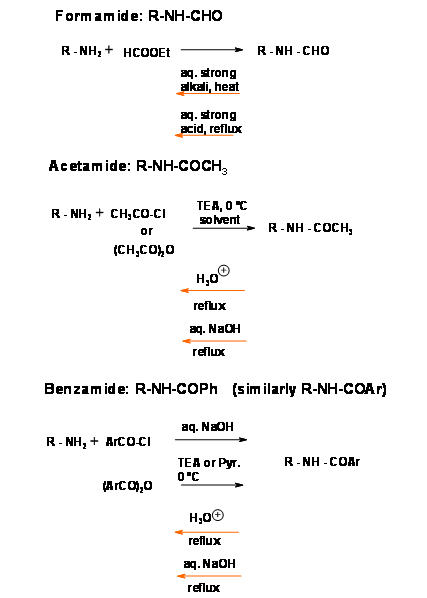
Protecciones de N-acetilo (N — COC H 3), N — Benzoilo (N — CoPH)
Estos son los grupos protectores clásicos para aminas primarias y secundarias. Los reactivos son baratos y el protocolo es sencillo. Tales amidas generalmente necesitan condiciones drásticas para la desprotección, aunque los rendimientos son generalmente buenos (Fig 4.1.10). Un procedimiento estándar es el reflujo en álcali acuoso o ácido mineral acuoso. Debido a las condiciones drásticas, se debe tener cuidado en este procedimiento para garantizar que se evite la racemización. Las amidas son generalmente sólidos cristalinos que se purifican fácilmente por cristalización. Cuando la protección se introduce en las primeras etapas de un esquema sintético largo y se desea una protección muy estable (como en la síntesis de nucleótidos) una amida es la protección más preferida.
Se han investigado varios enlaces amida más lábiles. Las amidas del ácido trifluoroacético son de especial interés. La introducción así como la escisión son simples y leves {Fig 4.1.11).
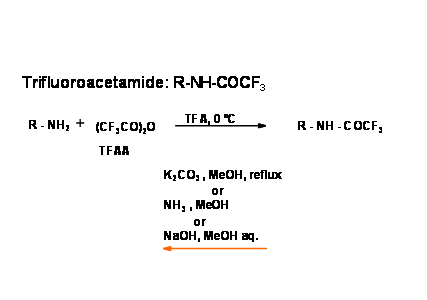
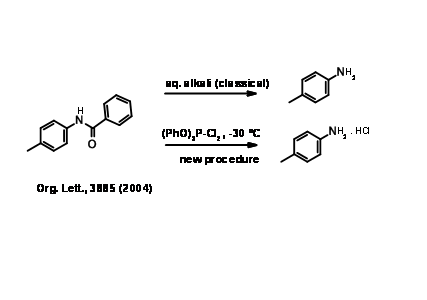
A continuación se da un reporte reciente sobre hidrólisis de amidas.
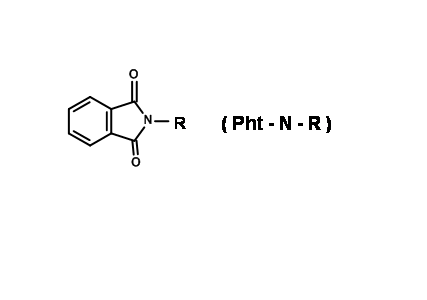
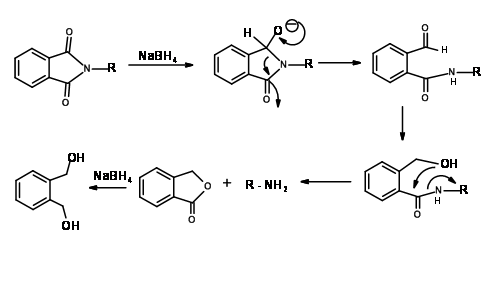
N — Protección contra ftaloilo (N — Pht)
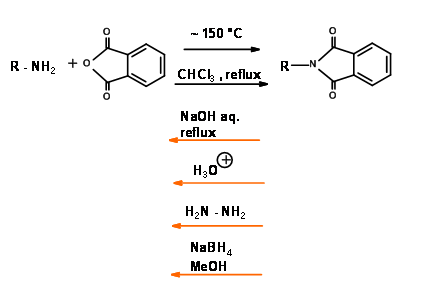
Mecanismo para NaB H 4 Reducción de N — Pht
N — Ésteres de ácido carboxílico como grupos protectores
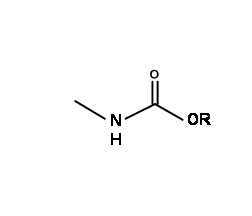
Como se describió anteriormente, los enlaces amida son muy fuertes. Por otro lado, los enlaces éster se escinden fácilmente por condiciones de base suaves. Una protección carboetoxi sobre amina tiene un enlace amida así como un enlace éster. Dado que los grupos N — COOH obtenidos en la hidrólisis son muy inestables, esta protección proporciona una gran familia de grupos protectores para aminas primarias y secundarias.
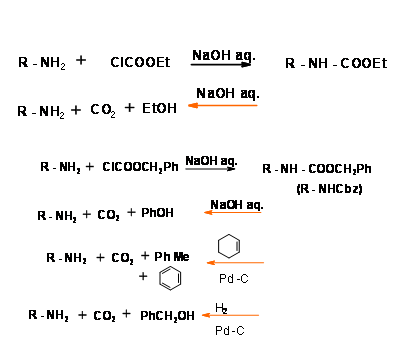
N — Carboetoxicarbonly (N — CoOEt) y Carbobenciloxicarbonilo (N — COOH2Ph) (N — Cbz o N — Z) Protecciones:
Estos grupos se introducen fácilmente utilizando los correspondientes ésteres de cloroformiato. Anhídridos o anhidridos mixtos en condiciones básicas suaves. Ambas protecciones podrían retirarse bajo agitación prolongada con base a temperatura ambiente. Aunque leve, a veces se observa alguna racemización. La protección N — Cbz tiene una ventaja añadida en que podría escindirse fácilmente en condiciones de hidrogenólisis (Fig 4.1.14). Sin embargo, la protección N — Cbz es estable a condiciones ácidas. Compare esto con la protección —Boc que se analiza a continuación.
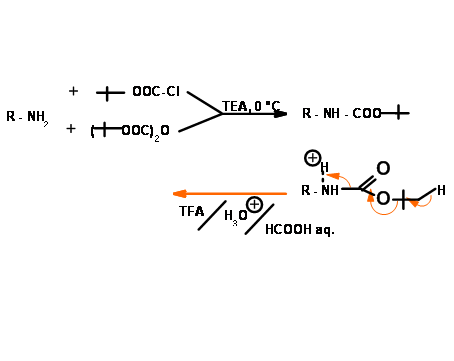
Protección con terc-butiloxicarbonil (N — CooBut, N — Boc)
La Protección Tert-Butiloxicarbonil podría introducirse y eliminarse en condiciones ácidas muy suaves. Esta protección es estable a álcalis e hidrogenólisis (Fig 4.1.15). Así, N — Z y N — Boc son complementarios como grupos protectores.
N — Protección Fluorometilenoxicarbonil (Fmoc)
Este grupo protector activo UV es muy popular en los protocolos de síntesis de péptidos en fase sólida (SPPS). Las etapas de protección y desprotección proceden en condiciones suaves y con buenos rendimientos (Fig 4.1.16).
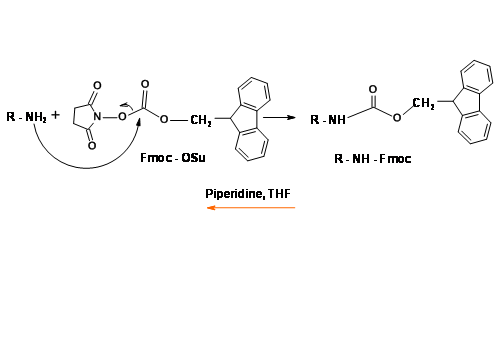
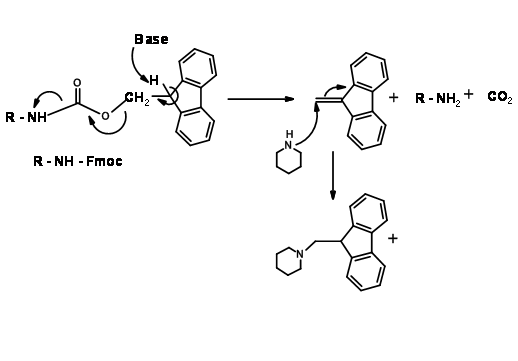
El mecanismo para la desprotección de Fmoc se muestra en (Fig. 4.1.17)
N — Sililación
La sililación es una protección común para el hidrógeno activo en heteroátomos. En el caso del enlace N — Si, los fluoruros de amonio cuaternario escinden este enlace (Fig 4.1.18).
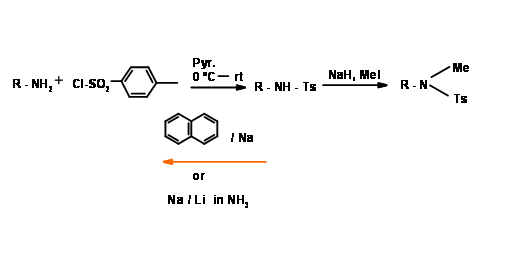
N — Tosilación (N — Tos)
Esta protección es muy estable. N — La tosilación se lleva a cabo fácilmente a través del procedimiento de cloruro de ácido. Se escinde por reacción de escisión electrónica solvatada. Cuando este grupo se une a una amina primaria, el grupo -NH se vuelve muy ácido (Fig 4.1.19).
Protección de — OH Groups
Acetatos (— Ac) y benzoatos (— Obz)):
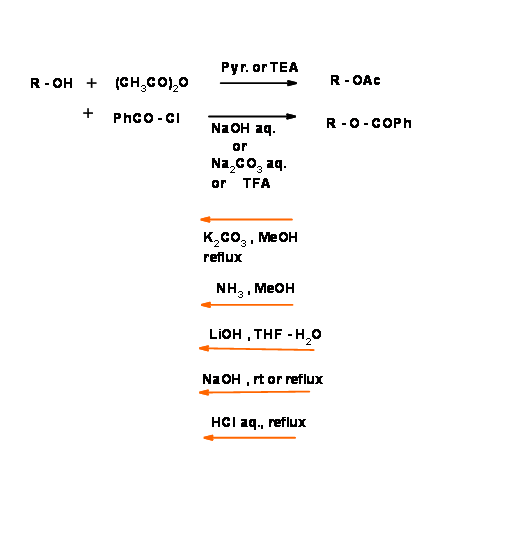
La química de protección del grupo — OH ha sido ampliamente investigada. La protección clásica es la formación de ésteres de ácidos carboxílicos alifáticos y aromáticos. Los ésteres aromáticos son comparativamente difíciles de hidrolizar en condiciones de base suave. Esto brinda una oportunidad para protocolos de desprotección selectiva (Fig. 4.1.20). Tenga en cuenta que esta protección es sensible tanto a las condiciones ácidas como a las bases.
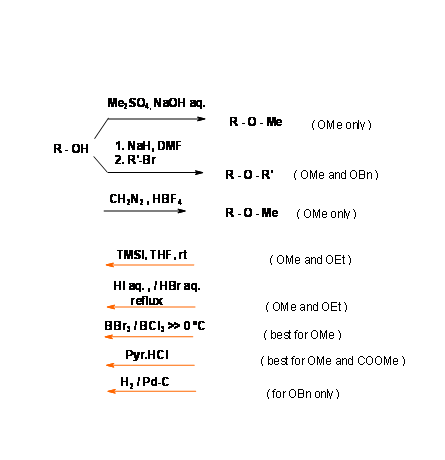
Éteres metílicos (— OMe) y bencil (R — OBn)
Un grupo éter es uno de los grupos funcionales más estables. De ahí que este grupo haya sido el grupo protector más favorecido. La desprotección fue un problema. A principios del siglo XX, el único procedimiento fue el reflujo con HI acuoso o HBr. En los últimos años han aparecido varios procedimientos nuevos para una remoción efectiva en condiciones leves. La característica especial de los bencil éteres es que esta protección se elimina fácilmente en condiciones neutras de hidrogenólisis (Fig 4.1.21). Sustituyentes como - OMe o - NO2 podrían introducirse en el anillo de benceno para modificar la reactividad en el sitio de protección.
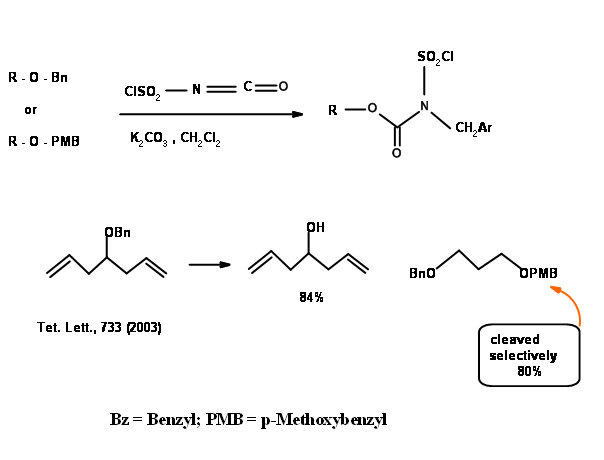
Cuando una olefina podría competir en el procedimiento de hidrogenólisis, la siguiente secuencia parece ser un procedimiento alternativo (Fig 4.1.22).
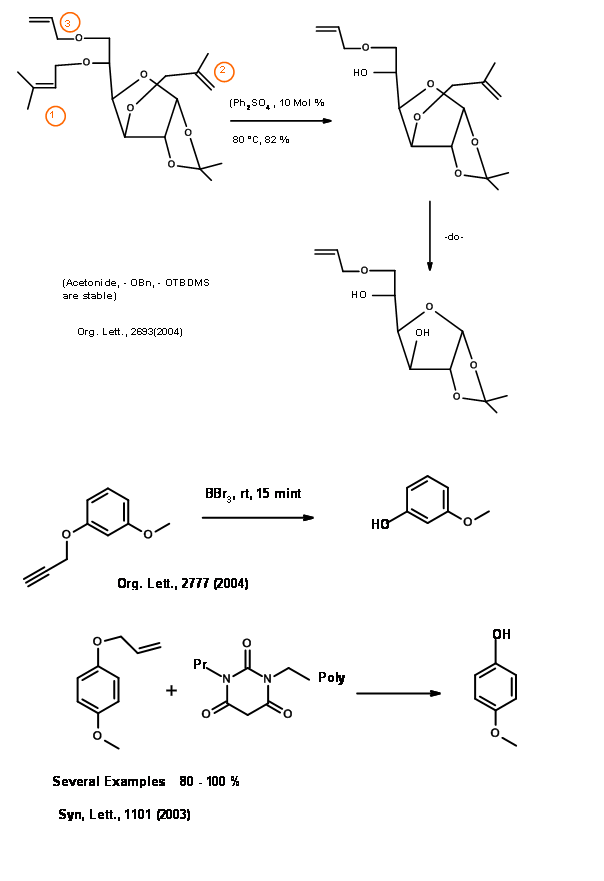
El éter alílico es una introducción reciente en la protección de — OH. La versatilidad de esta protección se pudo apreciar en los siguientes ejemplos (Fig 4.1.23).
Éteres de sililo (R — OSi R 3)
El enlace oxígeno-silicio sigma es estable a los reactivos de litio y Grignard, nucleófilos y reactivos de hidruro, pero muy inestable al agua y a las condiciones ácidas y bases acuosas suaves. Un silil éter de alcohol secundario es menos reactivo que el de un alcohol primario. El O — trimetilsililo (O — SiMe3) fue la primera protección de esta clase. (Fig 4.1.24).
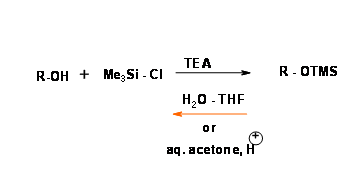
La sustitución del grupo metilo por otros grupos alquilo y arilo da una gran variedad de silil éter con diversos grados de estabilidad hacia la hidrólisis (Fig 4.1.25).

Los siguientes ejemplos ilustran la selectividad en la formación e hidrólisis de este grupo (Fig. 4.1.26).
Éter tetrahidropiranílico (— OTHP) y éter tetrahidrofuranílico (— OTHF)
Estos grupos protectores para los alcoholes son, de hecho, acetales. Se sintetizan utilizando el dihidropirano (DHP) y el dihidrofurano (DHF) respectivamente. Se comportan como acetales en su estabilidad y escisión (Fig 4.1.27). La tasa de formación y escisión para estos dos grupos difieren, lo que encuentra aplicación para la protección diferencial de alcoholes.
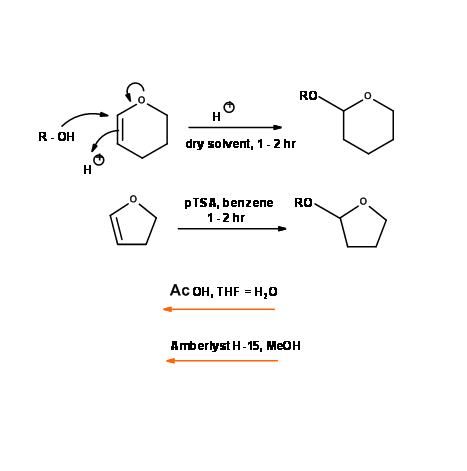
Estos grupos protectores encontraron un uso extensivo en la síntesis. Sin embargo, pronto se observaron dos inconvenientes importantes.
- Se genera un nuevo estereopunto al introducir esta protección. Aunque no es relevante desde el punto de vista de la molécula diana, en las moléculas quirales esto creó problemas de diastereómeros en espectroscopía (RMN y MS) y cromatografía.
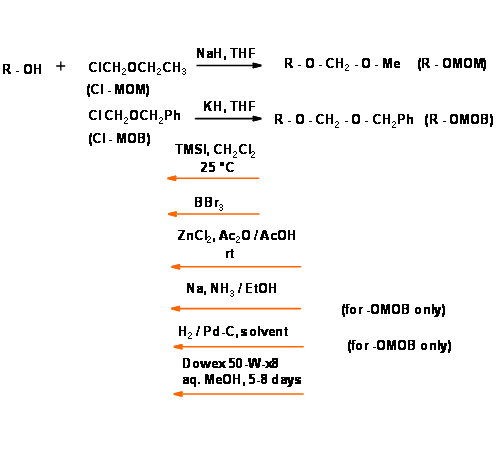
- Estos éteres ocasionalmente causaron explosiones en los procedimientos de hidroboración debido a la formación de peróxido. El problema diastereomérico se resolvió mediante la introducción de O-metilenoximetil éter (— O — MOM) y O — metilenoxibencil éter (R — O — MOB) (Fig 4.1.28). Varias otras modificaciones ya están disponibles.
Protección de vic — Diols
Al reaccionar con benzaldehído o acetona con un catalizador ácido adecuado, los vic-dioles forman acetales cíclicos. Esto de hecho es una prueba de la existencia de vic-dioles en la molécula. Son protecciones acetales y por lo tanto se comportan como acetales en su química (Fig 4.1.29)
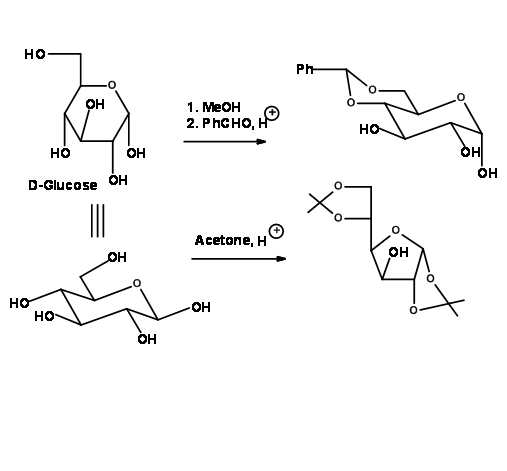
Conclusión
Las discusiones anteriores son solo un atisbo de la vasta literatura sobre este tema. Cuando hay más de un grupo funcional competidor presente en una molécula, puede ser necesario introducir al menos una etapa de protección y una desprotección en el esquema sintético. Esto se suma no sólo a la duración del esquema sintético, sino también al costo del compuesto final. Con la creciente conciencia en Química Verde, los químicos han estado tratando de reducir al mínimo este protocolo o preferiblemente evitarlo por completo. En la literatura se conocen varias síntesis libres de protección de productos naturales. Hablaríamos de este tema al final de este capítulo.
Lectura adicional
- Greene T. W., Grupos Protectores en Síntesis Orgánica. Wiley. N. Y., (1980), (1991).
- Smith M. B., Síntesis Orgánica, McGraw-Hill Inc, N. Y., (1994).
- Djerassi C., Reacciones con esteroides —Un esquema para químicos orgánicos, Holden-Day nc. San Francisco) 1963).
- Química Orgánica Avanzada: Principios Herramientas y Lógica de Síntesis, R. Balaji Rao, Vishal Publishing Co., Jalandhar, India (2012).
- Aminoácidos, Péptidos y Proteínas en Química Orgánica, Vol 4, Ed por Andrew B. Hughes (2011) Wiley-VCH; Protection Reaction, V.V. Sureshbabu y N. Narendra página 1 — 97.
4.2 Desconexión de bonos
Habiendo elegido la molécula TARGET para la síntesis, el siguiente ejercicio consiste en trazar planes sintéticos que resuman todas las rutas razonables para su síntesis. Durante las últimas décadas, los químicos han estado trabajando en un proceso llamado RETROSÍNTESIS. La retrosíntesis podría describirse como una desconexión lógica en los vínculos estratégicos de tal manera que el proceso conduciría progresivamente a materiales de partida fácilmente disponibles a través de varios planes sintéticos. Cada plan así evolucionado, describe una 'RUTA' basada en una retrosíntesis. Cada desconexión conduce a una estructura simplificada. La lógica de tales desconexiones forma la base para el retroanálisis de una molécula diana dada. Los productos naturales han proporcionado a los químicos una gran variedad de estructuras, teniendo funcionalidades complejas y estereoquímica. Esta área ha proporcionado varios objetivos desafiantes para el desarrollo de estos conceptos. El principio de subrayado en la concepción de enfoques lógicos para rutas sintéticas es muy similar al siguiente problema simple. Echemos un vistazo al siguiente bloque grande, que se realiza ensamblando varios bloques pequeños (Fig 4.2.1). Se podía ver fácilmente que el bloque grande se podía descomponer de diferentes maneras y luego volver a ensamblar para dar el mismo bloque original.
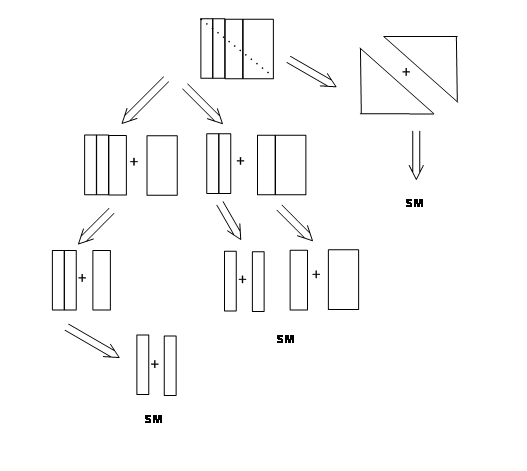
Ahora intentemos extender el mismo enfoque para la síntesis de una molécula simple. Analicemos tres posibles 'desconexiones' para un anillo de ciclohexano como se muestra en la Figura 4.2.2.
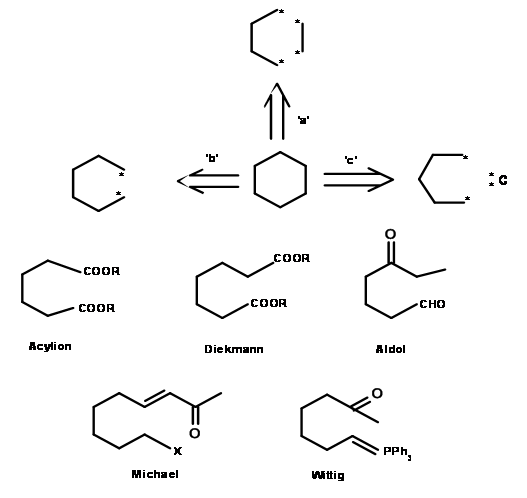
En el análisis anterior hemos intentado desarrollar tres formas de desconectar el anillo de seis miembros. ¿Hemos creado así tres vías para la síntesis del anillo de ciclohexano? ¿Tales desconexiones tienen sentido químico? Los antecedentes de un químico orgánico deberían permitirle leer el proceso como una reacción química en la dirección inversa (o 'retro-'). Los puntos en las estructuras anteriores podrían representar un ion carbonio, un carbanión, un radical libre o una reacción más compleja (como una reacción pericíclica o un reordenamiento). La aplicación de tal pensamiento químico podría abrir varias reacciones plausibles. Analicemos el camino b, que resultó de la escisión de un enlace sigma. Una ruta de ciclación aniónica sola expone varios candidatos como intermedios adecuados para la formación de este enlace. El análisis anterior describe solo tres caminos fuera del gran número de rutas de escisión alternativas que están disponibles. Un análisis extendido que se muestra a continuación indica más posibilidades de este tipo (Fig 4.2.3). Cada uno de estos intermedios podría ser sometido a un proceso de desconexión adicional y el proceso continuó hasta llegar a un material de partida razonablemente pequeño y fácilmente disponible. Así, se podría construir un 'ÁRBO-SINTÉTICO completo' que resumiría todas las rutas posibles para la molécula diana dada.
4.3 Eficiencia de una ruta
Se dice que una ruta es eficiente cuando el “rendimiento general” del proceso total es el mejor entre todas las rutas investigadas. Esto dependería no sólo del número de pasos involucrados en la síntesis, sino también del tipo de estrategia seguida. La estrategia podría implicar una “síntesis lineal” que implique solo pasos consecuentes o una “síntesis convergente” que implique menos pasos consecuentes. La Figura 4.3.1 que se muestra a continuación muestra algunos patrones que podrían reconocerse en dichos árboles sintéticos. Cuando cada proceso de desconexión conduce a un solo intermedio factible y el proceso avanza de esta manera
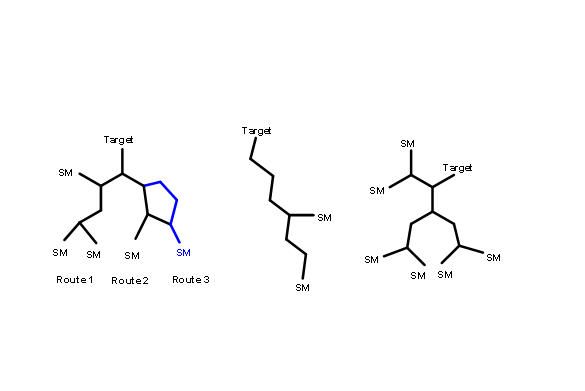
todo el camino a un conjunto de materiales de partida (SM), el proceso se llama Síntesis Lineal. Por otro lado, cuando un intermedio podría desconectarse de dos o más formas que conducen a diferentes intermedios, la ramificación ocurre en el plan. Los procesos podrían continuar hasta llegar a las SM. En tales rutas convergen diferentes ramas de las vías sintéticas hacia un intermedio. Tales esquemas se llaman Síntesis Convergente.
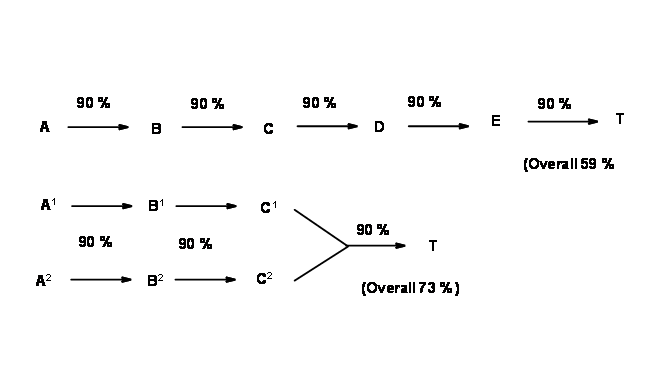
Los diagramas de flujo que se muestran a continuación (Fig. 4.3.2) representan una síntesis hipotética de 5 pasos por las dos estrategias anteriores. Asumiendo un rendimiento muy bueno (90%) en cada etapa (esto rara vez se ve en proyectos reales), una síntesis más liniana da 59% de rendimiento global, mientras que una síntesis convergente da 73% de rendimiento global para el mismo número de etapas.
4.4 Problema de los sustituyentes y estereoisómeros
La situación se vuelve más compleja cuando se considera la posibilidad de que se generen isómeros no deseados en diferentes etapas de la síntesis. El rendimiento general disminuye considerablemente para la síntesis del isómero correcto. Por lo tanto, se prefieren las reacciones que producen isómeros individuales (reacciones diastereoespecíficas) con buenos rendimientos. Algunas reacciones como la Reacción de Diels Alder generan varios estereopuntos (puntos en los que se generan los estereoisómeros) simultáneamente en un solo paso de una manera altamente predecible. Tales reacciones son muy valoradas en la planificación de estrategias sintéticas debido a que varias características estructurales deseables se introducen en un solo paso. Donde un enantiómero puro es el objetivo, la situación vuelve a ser compleja. Un compuesto puro en la etapa final aún podría tener 50% de enantiómero no deseado, lo que llevaría a una caída drástica en la eficiencia de la ruta. En tales casos, es deseable separar los isómeros ópticos lo antes posible en la ruta, a lo largo de la ruta sintética. Este es el mérito principal del Enfoque Quirón, en el que se elige el material de partida adecuado de una 'piscina quiral' barata y fácilmente disponible. Discutiremos este aspecto después de haber entendido la lógica de las síntesis de planeación. Dados estos parámetros, ahora podría decidir la ruta más eficiente para cualquier objetivo dado.
Las moléculas de interés suelen ser más complejas que el anillo simple de ciclohexano discutido anteriormente. Pueden tener sustituyentes y grupos funcionales en puntos especificados e incluso puntos estereoquímicos específicos. La construcción de un árbol sintético debería acomodar idealmente todos estos parámetros para dar rutas eficientes. Veamos un ejemplo un poco más complejo que se muestra en la Figura 4.4.1. La cetona 4.4.1A se requiere como intermedio en una síntesis. A diferencia del ciclohexano simple discutido anteriormente, el patrón de sustitución y el grupo ceto-en esta molécula imponen algunas restricciones a los procesos de desconexión.
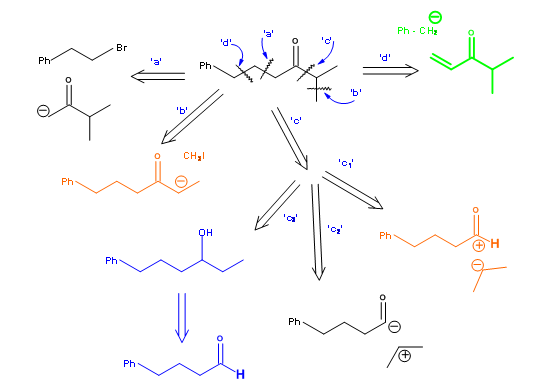
Escisión a: Esta vía implica el ataque de un anión de metilisopropilcetona sobre un componente bromo-componente. Escisión b: Esta ruta implica la metilación regioespecífica simple de una cetona más grande que porta todos los elementos estructurales restantes. Escisión c: Esta ruta implica tres posibilidades diferentes. La ruta C-1 contempla una unidad de acilonio, que podría provenir de un haluro de ácido o de un éster. La ruta C-2 implica una reacción umpolung en la unidad de acilo. La ruta C-3 sugiere una oxidación de un alcohol secundario, que podría obtenerse a través de una reacción tipo Grignard. Escisión d: Esto implica una adición Micheal.
Cada una de estas rutas podría desarrollarse aún más hacia atrás para completar el árbol sintético. Estas son solo algunas rutas plausibles para ilustrar un punto importante que los detalles sobre la estructura restringirían las posibles escisiones a algunos puntos estratégicos. Las contribuciones notables a la planificación de las síntesis orgánicas provinieron de la escuela de E.J. Corey. Estos desarrollos han sido compilados por Corey en un libro con el título Lógica DE SÍNTESIS QUÍMICA. Estas y varias presentaciones relacionadas sobre este tema deben tomarse como lineamientos. Se idean después de analizar la mayoría de los enfoques conocidos publicados en la literatura e identificar un patrón en la lógica. No es necesario que restrinjan el alcance de nuevas posibilidades. A continuación se describen algunas de las estrategias importantes.
4.5 Exploración preliminar
Cuando un químico sintético mira el Target dado, primero debe reflexionar sobre algunos pasos preliminares para simplificar el problema en cuestión. ¿La molécula es polimérica? Ver si toda la molécula podría dividirse en unidades monoméricas, las cuales podrían acoplarse mediante una reacción conocida. Esto se ve fácilmente en el caso de péptidos, nucleótidos y polímeros orgánicos. Esto también podría ser cierto para otros productos naturales. En moléculas como la C-Toxiferina 1 (4.5.1A) (Fig 4.5.1), el punto de dimerización es obvio. En varios otros casos, se requiere una visión más profunda para identificar las unidades monoméricas, como es el caso del ácido Usnico (4.5.1B). En el caso del antibiótico macrólido Nonactina (4.5.1C), esta estrategia reduce las posibilidades de síntesis de una unidad monomérica (4.5.1D). La estructura general tiene simetría S4 y es aquiral aunque ensamblada a partir de precursores quirales. Tanto el ácido (+) -nonáctico como el ácido (−) -nonáctico (4.5.1D) son necesarios para construir el macrociclo y se unen cabeza a cola en un patrón alterno (+) - (−) - (+) - (−). (véase J. Am. Chem. Soc., 131, 17155 (2009) y referencias allí citadas).

¿Una parte de la estructura ya está resuelta? El estudio crítico de la literatura a menudo puede revelar que la misma molécula o una estrechamente relacionada ha sido resuelta. R.B. Woodward sintetizó (4.5.2C) como intermedio clave en una elegante síntesis de Reserpina (4.5.2A). El mismo compuesto intermedio (4.5.2C) se convirtió en el compuesto de partida clave para Velluz et.al., en la síntesis de Deserpidina (4.5.2B) (Fig 4.5.2).
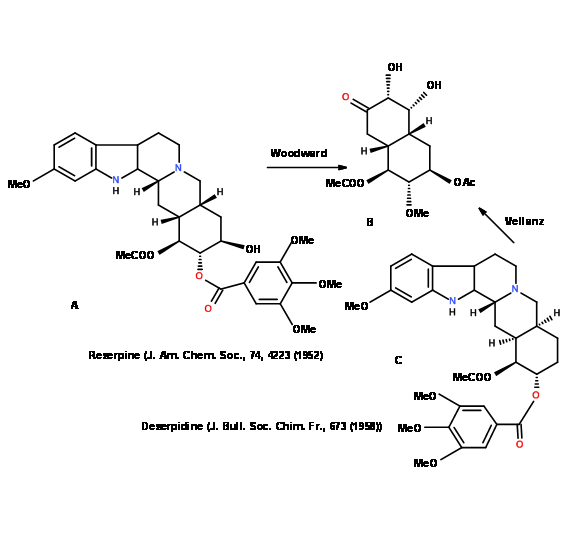
Dichas estrategias reducen el tiempo necesario para la síntesis de nuevos candidatos a fármacos. Estas estrategias se utilizan a menudo en la química de productos naturales y la química de fármacos. Una vez que se completa el escaneo preliminar, la molécula diana podría desconectarse en Bonos Estratégicos.
4.6 Vínculos estratégicos, retrones y transformadas
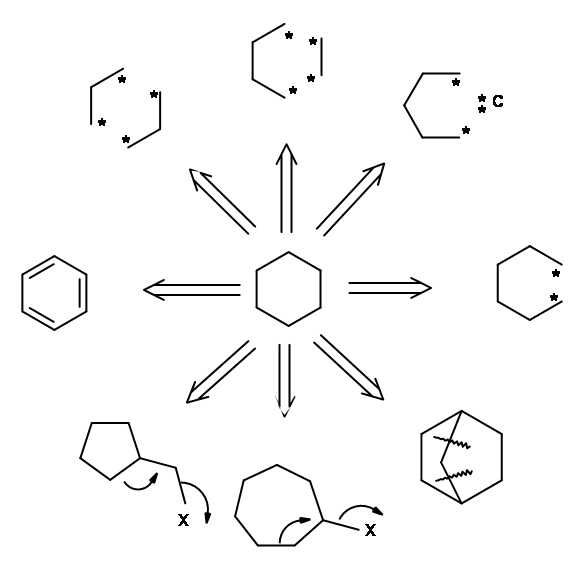
Los ENLAZOS ESTRATÉGICOS son los enlaces que se escinden para llegar a materiales de partida (SM) o SINTON Para fines de desconexión de enlaces, Corey ha sugerido que la estructura podría clasificarse de acuerdo con las subestructuras generadas por reacciones químicas conocidas. Llamó a las subestructuras RETRONS y las transformaciones químicas que generan estos Retrones se llamaron TRANSFORMAS. A continuación se presenta una breve lista de Transformadas y Retrones (TABLA 4.6.1). Tenga en cuenta que cuando las Transformadas generan retrones, el producto puede tener nuevos STEREPOINTS (detalles estereoquímicos) generados que pueden necesitar una valoración crítica.
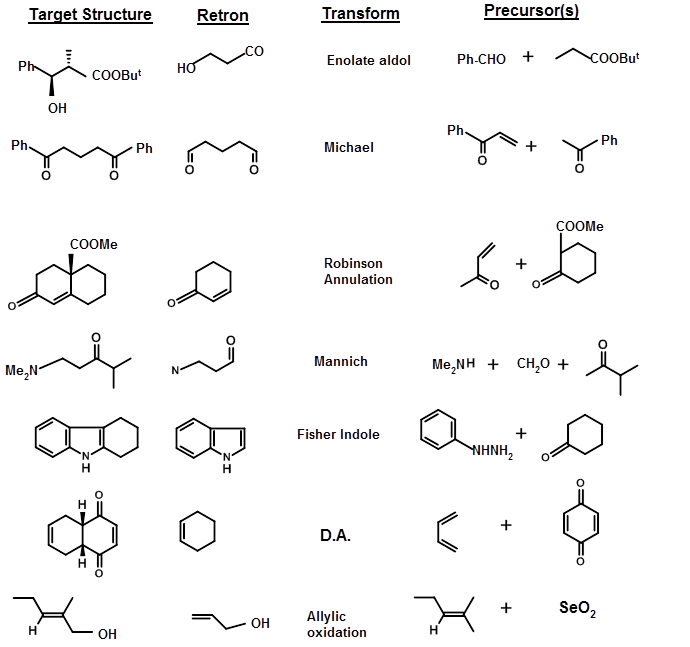
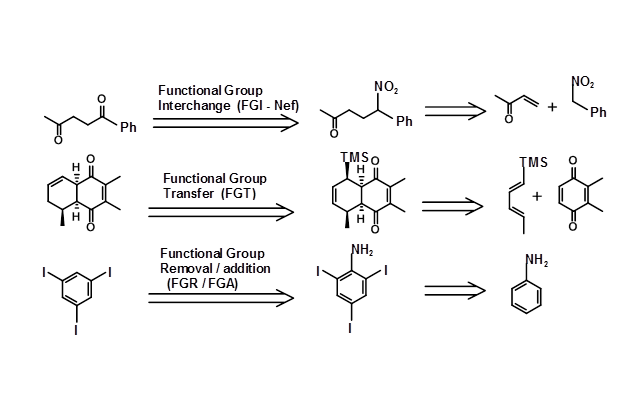
La estructura del objetivo podría ser tal que el Retron y las Transformadas correspondientes pudieran visualizarse fácilmente y aplicarse directamente. En algunos casos, los Transformados o los Retrones pueden no ser obvios. En varias síntesis, las transformaciones no simplifican la molécula, sino que facilitan el proceso de síntesis. Por ejemplo, se podría generar un grupo ceto-mediante la modificación de una unidad -CH-N O 2 a través de una reacción de Nef. Esto genera un nuevo conjunto de par Retron/transforma. Algunas de estas transformaciones se enumeran a continuación, junto con la nomenclatura sugerida por Corey (Fig 4.6.2).
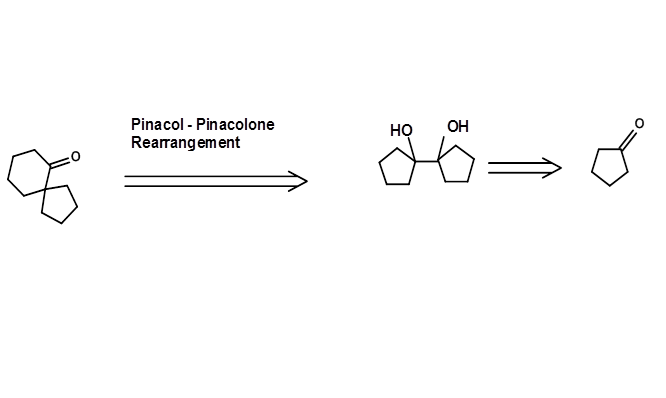
Una reacción de reordenamiento podría ser un método poderoso para generar nuevas subestructuras adecuadas. En el siguiente ejemplo, se obtiene un Pinacol Retron adecuado, necesario para el reordenamiento a través de una transformada de aciloína (Fig 4.6.3). Tal reordenamiento Los retrones a menudo no son obvios para ojos inexpertos.
Algunas transformaciones pueden ser necesarias para proteger (acetales para cetonas), modificar (reducción de una cetona a alcohol para evitar una condensación de Aldol durante una condensación de Claisen) o transponer un elemento estructural como un estereopunto (por ejemplo, inversión de S N 2, epimerización etc.,) o desplazar un grupo funcional. Tales transformadas no simplifican la unidad estructural dada. A veces, se puede introducir la activación en puntos específicos de la estructura para provocar la formación de un enlace C-C y posteriormente se puede eliminar el grupo extra. Por ejemplo, considere la siguiente retrosíntesis en la que se ha introducido un grupo éster extra para facilitar un Dieckmann Retron. En metas complejas, las combinaciones de tales estrategias podrían resultar una estrategia muy productiva en la planeación de la retrosíntesis. Sea testigo de la estrategia de modificación química que se muestra a continuación para una síntesis estereoespecífica eficiente de una olefina trisustituida (Fig. 4.6.4)
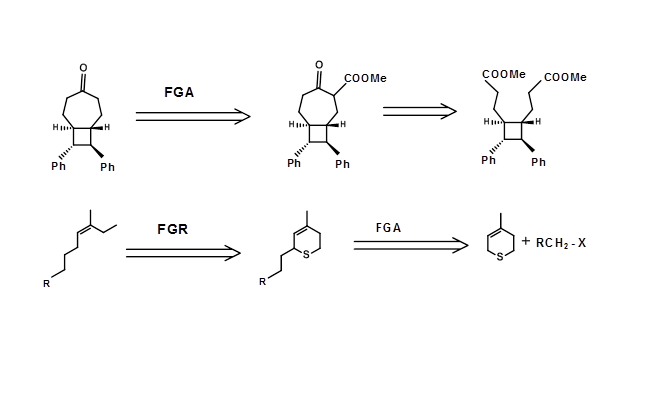
Figura 4.6.4 Ejemplos de estrategias FGA/FGR para objetivos complejos
Entre las arquitecturas moleculares, los anillos puenteados plantean un desafío complejo en los procedimientos de desconexión basados en estructuras. Corey ha sugerido pautas para desconexiones eficientes de bonos estratégicos.
Una escisión de enlaces para la retrosíntesis debe conducir a estructuras simplificadas, preferiblemente con anillos de cinco o seis miembros. Los anillos medianos y grandes son difíciles de sintetizar estereoespecíficamente. Entre los anillos comunes, un anillo de seis miembros es fácilmente abordado y manipulado a anillos grandes y pequeños. Escisión simultánea de dos enlaces, lo que sugiere cicloadición — los retrones suelen ser más eficientes. Algunas escisiones de enlaces estratégicos se muestran en la Figura 4.6.5, sugiriendo estrategias de escisión buenas y malas basadas en este enfoque. Sin embargo, estos lineamientos no son restrictivos.
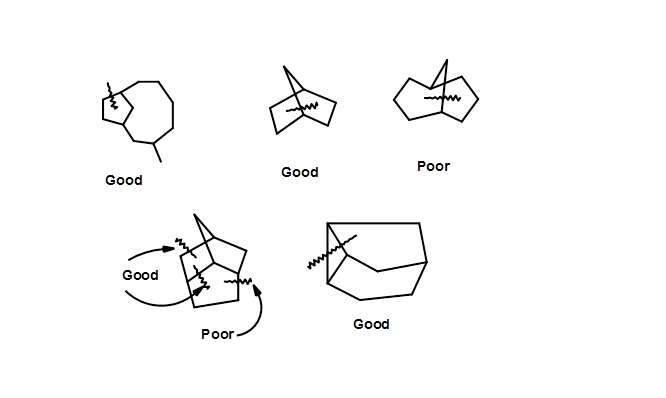
Identificar conjuntos de Retron — Transformar en una molécula diana dada es, por lo tanto, un componente crítico en la retrosíntesis. Tal enfoque a menudo podría generar varias rutas sintéticas. El mérito de este enfoque es que los materiales de partida no perjudican esta lógica. Las retrosíntesis así desarrolladas podrían abrir varias rutas que necesitan un mayor escrutinio crítico sobre la base de hechos conocidos.
La identificación de conjuntos de Retrons/Transforms proporcionó el requisito previo para programas asistidos por computadora diseñados para generar rutas retrosintéticas. Se entrelazaron una lista de Retrons y las transformaciones correspondientes y los datos se almacenaron en la computadora. Todas las reacciones conocidas fueron analizadas por sus características de Retron/Transform y documentadas. También se documentaron y vincularon las citas bibliográficas correspondientes. Con base en estos insumos, se diseñaron programas de computadora para generar rutas retrosintéticas para cualquier estructura dada. Varios de estos programas ya están disponibles en el mercado para ayudar a los químicos a generar estrategias sintéticas. Dada cualquier estructura, estos programas generan varias rutas. Una vez que el científico identifica las rutas específicas de interés para su posterior análisis, el programa genera pasos sintéticos detallados, reactivos requeridos y las citas correspondientes. A pesar de una inteligencia artificial tan poderosa, la inteligencia y el genio intuitivo de un químico siguen siendo capaces de generar una nueva estrategia, aún no programada. Nuevamente, la inteligencia humana sigue siendo un insumo crítico para el análisis de las rutas generadas usando una computadora. Con base en la experiencia del equipo de químicos, su objetivo proyectado del proyecto y las instalaciones disponibles, las rutas se proyectan aún más.
4.7 Elaboración de los conceptos
A continuación se presentan listas cortas de síntesis que ejemplifican estrategias de retroanálisis ideadas a través de poderosas transformaciones. Varias síntesis de la química de productos naturales se discuten más adelante en este capítulo, lo que ilustra aún más estos puntos.
Retrosíntesis basada en Transformada de Diels-Alder; (E.J. Corey et.al., J.A.C.S. (1972), 94, 2549). Fumagillol (4.7.1A) presenta 4 estereocentros y funcionalidades sensibles.
La simplificación de los grupos funcionales primero expuso un vic-diol. Este sitio podría provenir de una olefina D. La retroanlisis adicional condujo a una secuencia diana estructuralmente simplificada B a F. Un sistema de anillo de ciclohexeno es adecuado para una poderosa Transformada DA. Esta etapa generó dos estereocentros en una reacción y también una olefina en la posición correcta para la hidroxilación. El intermedio clave C también podría generarse a través de una transformación de grupo funcional que condujera a G. Esto proporcionó alcance para un nuevo conjunto de materiales de partida usando otra Transformada DA. El análisis retrosintético y la síntesis real se muestran en la Figura 4.7.1.
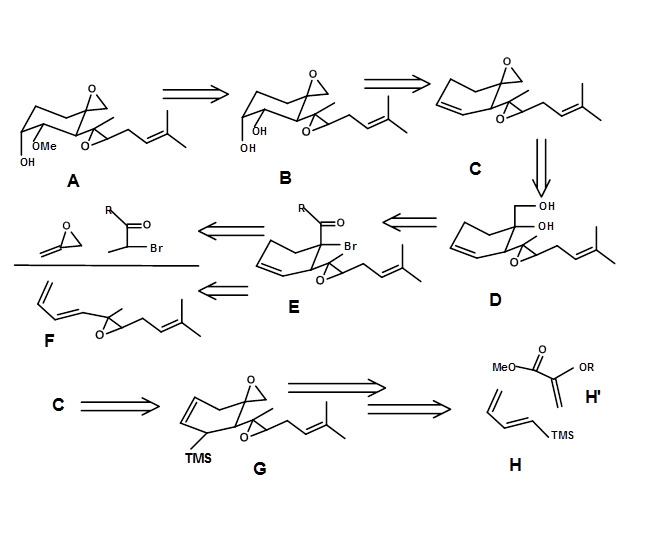
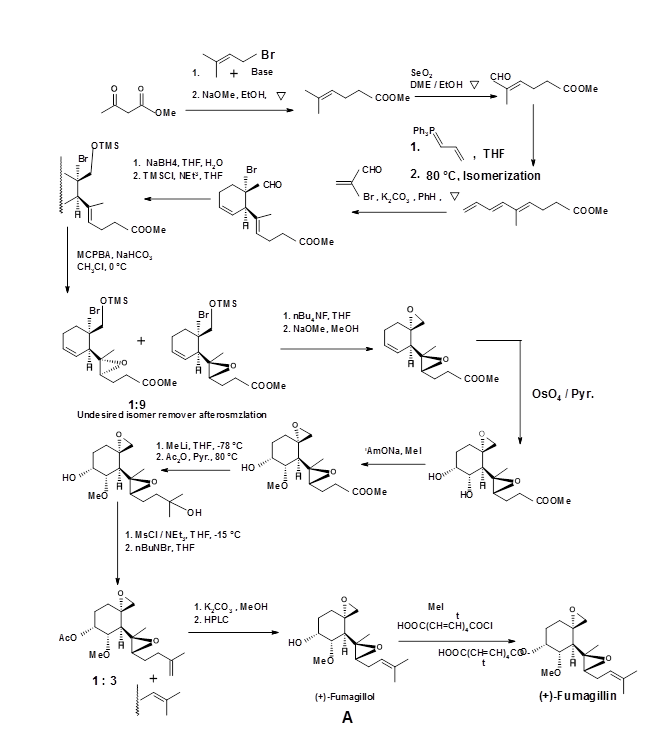
El protocolo sintético reportado por Corey se describe en la Figura 4.7.2
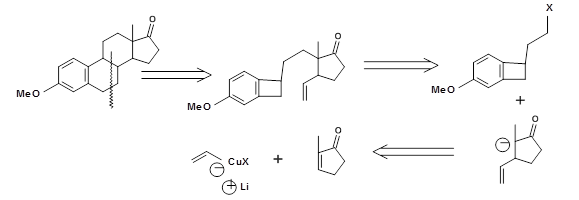
Para la síntesis de Estrone, Kametani et.al ideó una interesante estrategia DA Transform. La estrategia retrosintética se representa en la Fig 4.7.3. El precursor de dieno requerido se generó mediante reacción de cicloreversión de una unidad de ciclobuteno (T. Kametani et.al., Tetrahedron, (1981), 37, 3).
Las olefinas trisustituidas estereoespecíficas cruciales sobre escualeno (4.6.4B) se sintetizaron usando un Claisen Retron 4.7.4A (Fig 4.7.4). Obsérvese el enfoque doble de Claisen en esta estrategia.
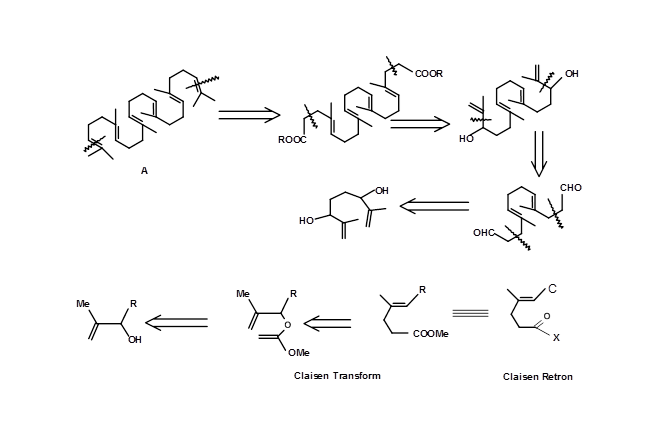
La ciclación de tipo biogenética de olefinas proporciona el alcance para la aplicación de Transformada Mecánica o Transformada basada en consideraciones mecanicistas. Una introducción con cuchillas de un centro quiral proporcionó una ruta eficiente para generar varios centros quirales enantiopuros en un solo paso usando esta estrategia (Fig 4.7.5).
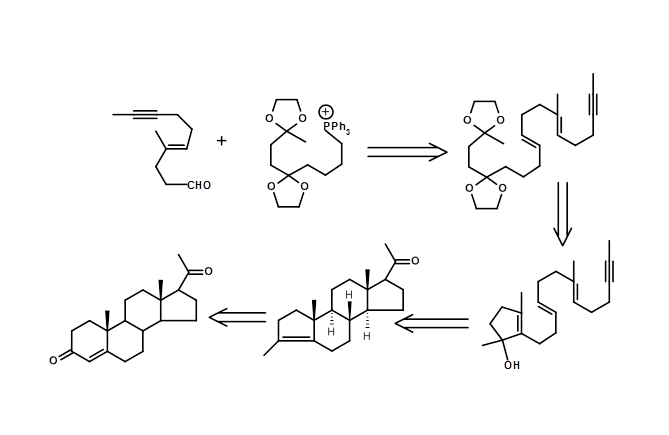
4.8 El problema de los enantiómeros
En estas largas discusiones anteriores, aprendimos acerca de los enfoques de desconexión. Dijimos que los estereocentros podrían introducir retos especiales en la planeación de rutas sintéticas eficientes. Veamos la molécula Biotina para entender las estrategias de desconexión y el problema de los estereocentros.
Baker estableció la estructura de Biotina en 1947 a través de una síntesis inequívoca de la molécula. Un retroanálisis del esquema sintético es como se muestra a continuación (Fig 4.8.1).
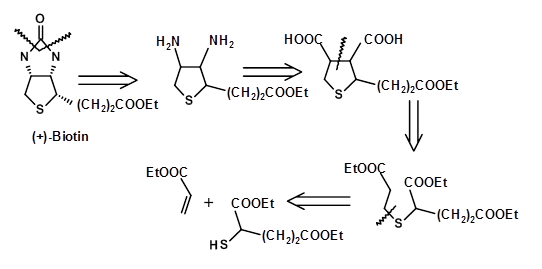
El SM elegido y el enfoque sintético establecieron claramente las conectividades atómicas y la estructura general del compuesto. Sin embargo, la ruta no intentó sintetizar un isómero puro debido a que la estereoquímica real no se estableció en ese momento. La ruta arrojó los ocho estereoisómeros (3 centros asimétricos). Estos isómeros se separaron cuidadosamente. En 1952 se identificó el isómero biológicamente activo como el enantiómero cis completo (+) -Biotoína. En esta etapa, varios grupos reportaron la síntesis estereoespecífica del isómero all cis exclusivamente (Fig 4.8.2). El siguiente retroanálisis describe tres intentos de este tipo. Nótese que estos esfuerzos se orientaron hacia la síntesis del racemato y no del isómero puro (+) de Biotina.
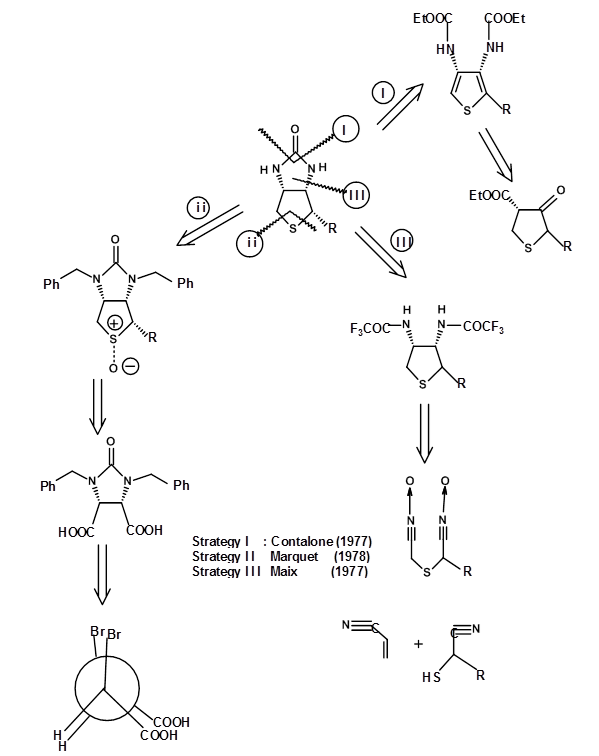
Estos enfoques resolvieron el problema de la pureza diastereomérica. Pero aún dejaron una mezcla de dos enentiómeros sin resolver a saber, (±) -Biotina. Para obtener un enantiómero puro con excelentes rendimientos, hay que resolver las mezclas racémicas en las etapas apropiadas. Como alternativa, se podría recurrir a la síntesis asimétrica en todas las etapas cruciales. Un enfoque aún mejor sería partir de un SM quiral, que tiene la mayoría de los estereocentros de la manera correcta. Este enfoque elegante se llama el enfoque Quirón. Cuando se ejecutan cuidadosamente, tales procedimientos producen enantiómero muy puro como producto final. Dos de estos enfoques para (+) -Biotina se muestran a continuación. En el primer enfoque se elige un aminoácido quiral cisteína porque tiene un centro asimétrico clave, el resto azufre y un ácido carboxílico en las posiciones correctas (Fig 4.8.3). En este ejemplo la elección del SM es bastante obvia. Obsérvese la introducción del segundo nitrógeno y la etapa de ciclación que conduce a la formación del anillo tetrahidrotiofeno. También tenga en cuenta que el rendimiento de los productos Cram vs anti-Cram (quelación) podría estar influenciado por la elección del reactivo. Este tipo de ideas vienen solo a través de un conocimiento profundo de esta reacción en particular.
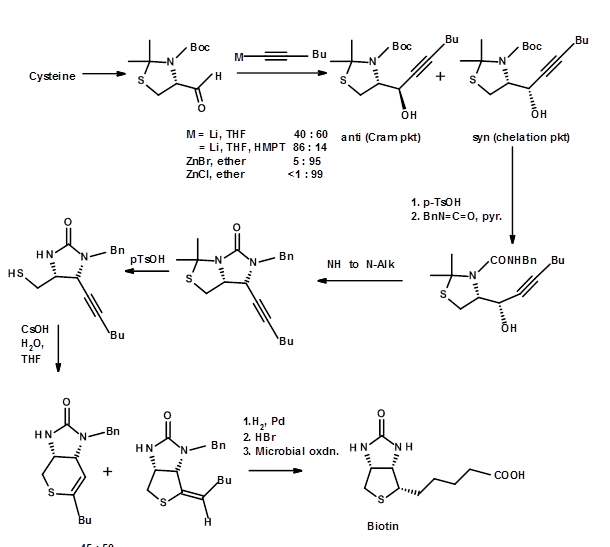
Fig. 4.8.3
En el segundo ejemplo aquí elegido, la elección del SM como el quirón apropiado no es obvia sino oculta. Tal análisis exige una visión más crítica de los estereocentros en cuestión.
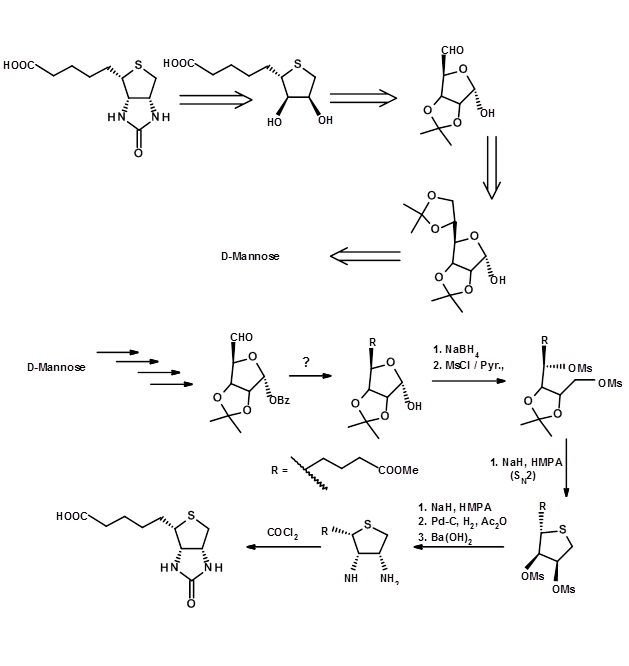
Conclusión
Un concepto emergente en la Lógica En Síntesis es la planeación deliberada de Caminos Sintéticos Verdes. La lógica del retroanálisis es la misma que se discutió anteriormente. El único punto diferenciador es que los criterios de selección de ruta sintética discutidos anteriormente analizarían ahora el mismo árbol sintético a través de una ventana de Química Verde para seleccionar solo aquellas rutas que tengan aspectos verdes máximos. El objetivo de la química verde se aplica a través de la inclusión de los Doce Principios de Química Verde. Esto podría hacerse adoptando una o más de las siguientes técnicas - Uso de Fuentes de Energía Verde como Microondas, Sonoquímica, Fotoquímica etc., síntesis sin disolventes, utilizando nuevos solventes fácilmente recuperables y solventes ecológicos, catalizadores reutilizables en síntesis y esquemas que evitan el grupo protector química. La mayor parte de la química utilizada en la Química Verde no es realmente nueva para los químicos. La química ahora es revisitada debido a la conciencia ambiental que ahora se ha deslizado en la química industrial y la sociedad en general. La mayor parte de la química está enterrada en dos siglos de literatura química. Varios nuevos descubrimientos en reactivos han aparecido en los últimos años. Ahora los químicos tienen que estar más alertas ante este despertar ante los daños ambientales causados por las actividades químicas en este globo.
Las discusiones anteriores solo tienen por objeto ilustrar los principales pasos involucrados en el análisis retrosintético de una molécula. El conocimiento profundo de las herramientas sintéticas, los mecanismos y la estereoquímica son requisitos previos esenciales para que un químico se aventure en la síntesis de moléculas complejas. No hace falta agregar que todos estos esfuerzos tienen que ser debidamente respaldados por un equipo de químicos, teniendo una rigurosa formación en técnicas de laboratorio, una experiencia de primera mano en varias reacciones/reactivos orgánicos y un profundo conocimiento de técnicas de purificación, técnicas espectroscópicas y no menos importante, un buen conocimiento de técnicas de búsqueda para escanear y recuperar la información requerida de la vasta literatura química acumulada desde los albores de la química moderna.
Retroanálisis de algunas moléculas interesantes
Ahora profundicemos en algunas estructuras seleccionadas elegidas de la química de productos naturales y veamos cómo se han abordado estas estructuras a través de diferentes estrategias sintéticas. Comenzaríamos con una molécula simple —Disparlure— con sólo dos centros asimétricos. El curso terminaría con un flovour de algunas síntesis basadas en Química Verde para llamar la atención de los estudiantes sobre estos conceptos e inquietudes recién emergentes.